






To show how to generate a consensus sequence from the information of massive parallel sequences data obtained from routine HIV anti-retroviral resistance studies, and that may be suitable for molecular epidemiology studies.
Material and methodsPaired Sanger (Trugene-Siemens) and next-generation sequencing (NGS) (454 GSJunior-Roche) HIV RT and protease sequences from 62 patients were studied. NGS consensus sequences were generated using Mesquite, using 10%, 15%, and 20% thresholds. Molecular evolutionary genetics analysis (MEGA) was used for phylogenetic studies.
ResultsAt a 10% threshold, NGS-Sanger sequences from 17/62 patients were phylogenetically related, with a median bootstrap-value of 88% (IQR 83.5–95.5). Association increased to 36/62 sequences, median bootstrap 94% (IQR 85.5–98), using a 15% threshold. Maximum association was at the 20% threshold, with 61/62 sequences associated, and a median bootstrap value of 99% (IQR 98–100).
ConclusionA safe method is presented to generate consensus sequences from HIV-NGS data at 20% threshold, which will prove useful for molecular epidemiological studies.
Generar una secuencia consenso a partir de los datos de secuenciación masiva obtenidos en estudios de resistencias a antiretrovirales, que sea representativa de la secuencia Sanger y que sirva para estudios de epidemiología molecular.
Material y métodosEn 62 pacientes se obtuvo la secuencia de transcriptasa reversa-proteasa, mediante Sanger (Trugene-Siemens), y NGS (454GSJunior-Roche). Las secuencias consenso NGS se generaron con Mesquite, seleccionando umbrales 10%, 15% y 20%. Para el estudio filogenético se empleó MEGA.
ResultadosUtilizando el umbral 10%, 17/62 pacientes presentaron secuencias pareadas NGS-Sanger, con una mediana de bootstrap del 88% (IQR 83,5-95,5). La asociación aumenta a 36/62 pacientes y el bootstrap, a 94% (IQR 85,5-98), y alcanza el máximo al 20% en 61/62 pacientes, bootstrap 99% (IQR 98-100).
ConclusiónMostramos un método seguro para generar secuencias consenso NGS para su uso en estudios de epidemiología molecular procesadas con umbral 20%, de fácil uso y aplicación en los servicios de microbiología clínica.
Many clinical microbiology departments have started to use next generation sequencing (NGS) techniques to study antiretroviral resistance in HIV patients. Several studies1 have shown the capacity of NGS to detect low abundance viral variants by lowering sensitivity below the 1% threshold (minority variants). This can greatly improve therapeutic decision-making and prevent therapeutic failure.2,3 In Spain, the use of NGS for the detection of antiretroviral resistance has been prompted to a certain extent by the decision of some suppliers to discontinue supply of Sanger sequencing systems.
Protease (PR) and reverse transcriptase (RT) sequences obtained from drug resistance tests are often used by researchers in molecular epidemiology studies, using phylogenetic and phylodynamic techniques.4 With the introduction of NGS techniques, this information can be lost due the complexity of processing and storing the sequences for this type of study; in addition, incorrect processing of NGS sequences can yield incorrect results. Special training in sequence processing and high-performance computers capable of processing the vast amounts of data generated are needed in order to use NGS sequences in phylogenetic studies.5 An alternative, in the case of molecular epidemiology studies, is to generate a single consensus NGS sequence; however, some studies do not clearly describe, or omit altogether, the method used to generate the sequence.6 In addition, we cannot know for certain the extent to which this consensus NGS represents the sequence obtained by Sanger methods and the effect of the thresholds used to generate this consensus.
The objective of this study was to determine the best threshold for obtaining a consensus NGS sequence that is representative of the Sanger sequence and can be used in molecular epidemiology studies.
MethodsFor the purpose of our study, we used sequences from 62 treatment naïve patients, newly diagnosed with HIV between 2014 and 2015 and referred for antiretroviral resistance studies. Sanger sequences were obtained using Trugene® HIV-1 Genotyping (Siemens – [NAD]). For NGS, we used the GSV-type HIV-1 Drug Resistance Primer kit (Roche) for 454 GS-Junior, based on the same RNA. The NGS consensus sequences are generated using the Mesquite v. 2.75 software, setting thresholds at 10%, 15% and 20%. Prior to the use of Mesquite, the sequences were filtered using Usearch fastq_filter commands, according to the desired amplicon length and sequence quality (>30 Q). Mesquite7 is a programme with an intuitive icon- and tab-based interface. Before processing data on Mesquite, the filtered sequences must be exported in pfam format and the threshold for the consensus sequence must be set before exporting in fasta format. Following this, the pol gene sequences (PR 4–99; RT 38–247) are processed, aligned by MUSCLE in MEGA 6.06 and phylogenetic trees are generated using the maximum likelihood method, using the General Time Reversible (GTR) model to calculate evolutionary distances, with a gamma distribution equivalent to 1.89 obtained with FindModel DNA, and using bootstrap resampling with 1000 replicas to build the consensus phylogenetic trees. To define a relationship between sequences, only the branches from clusters with a bootstrap value greater than 75% are taken into consideration. Finally, the trees are processed in FigTree v. 1.4.2. The viral subtype analysis was performed using the REGA HIV-1 Subtyping Tool v. 3.0.
ResultsOur study included 62 treatment naïve HIV-1 patients with a median age of 37 years (IQR 30–45), viral load (median) of 74,900cp/ml (IQR 20,715–176,250), CD4 count (median) of 430cells/ml (IQR 48.5–567.78); 82% were men.
To evaluate concordance between the NGS consensus sequences with different thresholds and the original Sanger sequence, we analysed the number of sequences with inter-related pairs and the bootstrap values between the pairs. Using a 10% threshold, we observed that the Sanger sequence pairs were correlated with NGS from the same sample in only in 17/62 (27%) patients and in these, the median bootstrap value was 88% (IQR 83.5–95.5). Increasing the threshold to 15%, the sequence pairs were correlated in 36/62 (58%) patients, with a median bootstrap value of 94% (IQR 85.5–98). At 20%, sequences were correlated in 61/62 patients, with a median bootstrap value of 99% (IQR 98–100) (Fig. 1). A large number of differences between base pairs were detected in cases where the NGS sequence was not correlated with the Sanger sequence.
 NGS-10%; (B) NGS-15%, and (C) NGS-20%. The bootstrap values are shown according to colour. A good association is 70% and over.")
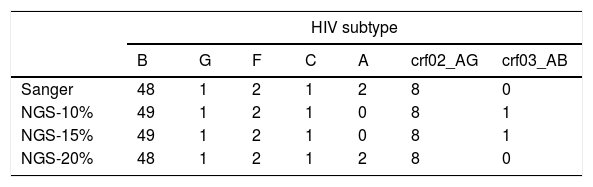
Most patients were infected by subtype B (77.4%), followed by CRF02_AG (12.9%), A and F (3.2%) and C and G (1.6%). Using a consensus NGS threshold of 10% and 15%, we observed 2 cases that differed from the Sanger subtype: one case of subtype B-NGS and A1-Sanger and another one from subtype CRF03_AB-NGS and A1-Sanger. These differences disappeared when using the 20% threshold consensus NGS sequences (Table 1). Fig. 2 shows the bootscan plot of the subtype in the second discordant sample.
, NGS-10% consensus sequence (B) and NGS-20% consensus sequence (C), using the REGA HIV-1 Subtyping Tool v. 3.0. In the Bootscan, the HIV A subtype value is the same in the Sanger and NGS-20% consensus sequence, however, a CRF03_AB subtype can be seen in the NGS-10% consensus sequence.")
Bootscan of the Sanger sequence (A), NGS-10% consensus sequence (B) and NGS-20% consensus sequence (C), using the REGA HIV-1 Subtyping Tool v. 3.0. In the Bootscan, the HIV A subtype value is the same in the Sanger and NGS-20% consensus sequence, however, a CRF03_AB subtype can be seen in the NGS-10% consensus sequence.
Phylogenetic studies of HIV,8,9 specifically studies into relatedness, transmission dynamics of the HIV epidemic and molecular pol gene sequence subtyping, have been used for various purposes, among them to understand HIV transmission networks and clusters and the migratory networks of the different subtypes. Most studies published at the international10 and local level11,12 have used Sanger sequencing. Some of these studies have used all the information obtained through NGS13 but investigators generally try to generate a single consensus sequence, usually using complex computer commands. The transition from Sanger sequencing to NGS for pol gene analysis of resistance mutations has changed the type of sequences used in clinical microbiology departments and can, paradoxically, stand in the way of local HIV molecular epidemiology studies in Spain. In this study, we propose using Mesquite, an intuitive, user-friendly, software without the need for commands that simplifies the process of obtaining a consensus sequence from NGS-generated sequences. We have shown that using a threshold of 20% to generate this consensus yields safe, reliable information that is identical to that obtained using Sanger sequencing. This information can be used in molecular epidemiology studies and solves the current problems arising from the use of NGS sequences.
As we have shown in this study, the threshold must be raised to 20% in order to safely use consensus sequences that are representative of Sanger sequences in HIV molecular epidemiology studies. This was the only threshold that yields a median bootstrap value of 99% (IQR 98–100) between the NGS consensus sequences and the Sanger sequence. Using thresholds of 10% or 15%, the percentage of correlated NGS-Sanger sequence pairs and the median bootstrap values are too low. Because of this variability, errors are made even in the determination of the viral subtype; this was corrected with the 20% consensus. These discrepancies are due to the multitude of ambiguous base pairs generated with the 10% and 15% thresholds, which made it impossible to correctly determine the viral subtype.
An important part of molecular epidemiology studies is sequence alignment, in which homologous positions are aligned based on the true evolutionary history of the sequences.14 The problem with using 10% and 15% NGS consensus sequences in such studies lies in the presence of ambiguous regions, which present substantial uncertainty and detract from the robustness of both phylogenetic15 and subtype statistical analyses, yielding unexpected results.
It is important to point out that the methodology presented here is appropriate for obtaining consensus sequences for use in HIV molecular epidemiology studies but not for the analysis of resistance mutations. The greater sensitivity of NGS to detect minority variants and its clinical utility have been studied in detail.1–4 NGS provides very valuable information on the relative proportion of a mutation with respect to the total circulating viruses. This information would be lost when obtaining the consensus sequence.
In summary, we present a methodology for generating consensus sequences that are representative of the Sanger sequence for use in molecular epidemiology studies by processing sequences with a threshold of at least 20%.
FundingHealth Research Fund (PI12/01053, PI15/00713), RD12/0017/006 (National R+D+I Programme, European Regional Development Fund—ERDF). Federico García is part of a Research Intensification Programme from the Andalusian Health Service. José Ángel Fernández-Caballero has an RD12/0017/006 contract.
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Fernández-Caballero Rico JÁ, Chueca Porcuna N, Álvarez Estévez M, Mosquera Gutiérrez MM, Marcos Maeso MÁ, García F. Validación de un método seguro y sencillo para la elaboración de secuencias consenso del virus de la inmunodeficiencia humana a partir de los datos de secuenciación masiva 454. Enferm Infecc Microbiol Clin. 2018;36:91–94.
