





The emergence of multiple variants of SARS-CoV-2 during the COVID-19 pandemic is of great world concern. Until now, their analysis has mainly focused on next-generation sequencing. However, this technique is expensive and requires sophisticated equipment, long processing times, and highly qualified technical personnel with experience in bioinformatics. To contribute to the analysis of variants of interest and variants of concern, increase the diagnostic capacity, and process samples to carry out genomic surveillance, we propose a quick and easy methodology to apply, based on Sanger sequencing of 3 gene fragments that code for protein spike.
MethodsFifteen positive samples for SARS-CoV-2 with a cycle threshold below 25 were sequenced by Sanger and next-generation sequencing methodologies. The data obtained were analyzed on the Nextstrain and PANGO Lineages platforms.
ResultsBoth methodologies allowed the identification of the variants of interest reported by the WHO. Two samples were identified as Alpha, 3 Gamma, one Delta, 3 Mu, one Omicron, and 5 strains were close to the initial Wuhan-Hu-1 virus isolate. According to in silico analysis, key mutations can also be detected to identify and classify other variants not evaluated in the study.
ConclusionThe different SARS-CoV-2 lineages of interest and concern are classified quickly, agilely, and reliably with the Sanger sequencing methodology.
La aparición de múltiples variantes del SARS-CoV-2 durante la pandemia de COVID-19 es motivo de gran preocupación mundial. Hasta el momento, su análisis se ha centrado principalmente en la secuenciación de nueva generación. Sin embargo, esta técnica es costosa y requiere equipos sofisticados, largos tiempos de procesamiento y personal técnico altamente cualificado con experiencia en bioinformática. Para contribuir al análisis de variantes de interés y de preocupación, aumentar la capacidad diagnóstica y procesar muestras para realizar vigilancia genómica, proponemos una metodología rápida y fácil de aplicar, basada en la secuenciación Sanger de 3 fragmentos del gen que codifica para la proteína espiga.
MétodosSe secuenciaron 15 muestras positivas para SARS-CoV-2 con un valor de umbral de ciclo inferior a 25 por metodologías Sanger y secuenciación de nueva generación. Los datos obtenidos fueron analizados en las plataformas Nextstrain y PANGO Lineages.
ResultadosAmbas metodologías permitieron identificar las variantes de interés reportadas por la OMS. Se identificaron 2 muestras como alfa, 3 gamma, una delta, tres mu, una ómicron y 5 cepas cercanas al aislado inicial del virus Wuhan-Hu-1. Según el análisis in silico, también se pueden detectar mutaciones clave para identificar y clasificar otras variantes no evaluadas en el estudio.
ConclusiónLos diferentes linajes de interés y preocupación de SARS-CoV-2 se clasifican de forma rápida, ágil y fiable con la metodología de secuenciación de Sanger.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a coronavirus that emerged in 2019 and is responsible for the current pandemic known as coronavirus disease 2019 (COVID-19)1. COVID-19 has since become a global public health threat, causing approximately 405 million infections and 5.8 million deaths worldwide2.
Early diagnosis is crucial to controlling the spread of COVID-19. The molecular detection of SARS-CoV-2 nucleic acid by real-time polymerase chain reaction (PCR) is considered the gold standard. However, it can be affected by virus mutations in the 5′(UTR), ORF1ab, spike (S), membrane, and nucleocapsid regions3,4, which favour an increase in the rate of false negatives5.
Mutations in the SARS-CoV-2 genome generate changes in protein structures, affecting their virulence, transmissibility and antigenicity6,7. Therefore, genomic surveillance of the virus has been essential in detecting early variants of interest (VOI) or variants of concern (VOC). All variants have mutations in the protein S4 gene. Therefore, most therapeutic strategies and vaccine development have focused on this gene.
The detection of the different mutations and the typification of the variants is carried out by next-generation sequencing (NGS) of the complete SARS-CoV-2 genome. The virus sequences are deposited in the Global Initiative on Sharing All Influenza Data EpiCoV™ database, so that genetic information from positive samples for SARS-CoV-28 can be shared. Similarly, there are platforms that enable genomic and phylogenetic analyses of the variants, such as Nextstrain9.
Although NGS techniques are crucial for molecular surveillance, they require a high degree of technical expertise, knowledge of bioinformatics and access to expensive equipment, rendering widespread use difficult. For these reasons, other methodologies have been designed, such as Sanger sequencing and real-time PCR, as variant screening tests3,10.
In Colombia, SARS-CoV-2 genomic characterisation and surveillance activities are centralised in the National Institute of Health. However, with the aim of contributing to genomic surveillance, increasing diagnostic capacity and simple workflow, and improving variant tracking in the shortest possible time, we propose a method based on Sanger sequencing of three fragments that partially cover the gene encoding protein S to detect VOI and VOC.
MethodsGenomic target selection and primer designThe reference sequence of SARS-CoV-2, GenBank: NC_045512, and the NCBI-Primer BLAST software (https://www.ncbi.nlm.nih.gov/tools/primer-blast/, https://www.ncbi.nlm.nih.gov/genbank/) was used to design the primers. The quality of the oligonucleotide design was evaluated by OligoAnalyzer v3.1 (https://www.idtDNA.com/calc/analyzer). Primer specificity was verified by in silico predictive analysis with the online Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
In silico simulation of detection of variants of interest and concernThe possible mutations in the amino acid sequence of the GenBank protein S were simulated. The reference sequences of the VOC (alpha, beta, gamma, delta and omicron) and VOI (lambda and mu) were obtained. They were aligned with the Sequencher 5.4.6 program (Gene Codes Corporation) and the variants were identified with the Nextstrain–Nextclade Web 1.10.0 web-based program, Nextclade CLI 1.7.0 (09-12-2021) (https://clades.nextstrain.org/).
SARS-CoV-2 positive patient samplesFor the screening of SARS-CoV-2 variants, 15 clinical samples from patients in Medellín (Colombia) with a positive result by real-time PCR for COVID-19 between 2020 and 2021 with a cycle threshold value under 25 were selected. The samples were processed first using the modified, standardised and validated Charité-Berlin protocol at the Laboratorio Integrado de Medicina Especializada11. The samples were taken following the guidelines of the Ministry of Health and Social Protection12.
RNA extraction and cDNA synthesisRNA was obtained using automated extraction systems with magnet technology, KingFisher™ Duo Prime System (Thermo Fisher Scientific Inc., USA.) with the MagMAX™ Viral/Pathogen Nucleic Isolation kit (Thermo Fisher Scientific Inc., USA). Similarly, manual extractions of some samples were carried out with the Quick-RNA Viral Kit (Zymo Research, USA). Total RNA was converted to cDNA using the TruScript™ First Strand cDNA Synthesis kit (Norgen, Thorold, Canada), following the manufacturer’s recommendations.
Conventional PCR for the detection of SARS-CoV-2 fragmentsThree independent standard PCRs were performed. The reaction conditions were: 2 μl Taq DNA Polymerase PCR Buffer 10X (Invitrogen, USA), 1.2 μl MgCl2 50 mM (ThermoFisher Scientific Inc., USA), 2 μl dNTP Mix 10 mM (Thermo Fisher Scientific Inc., UU.), 0.3 μl sense and antisense primers 20 μM, 0.16 μl Platinum Taq DNA Polymerase (Invitrogen, USA) and 2 μl de cDNA for a final volume of 20 μl per sample.
The thermocycling conditions were: 94 °C × 5 min, followed by 35 cycles at 94 °C × 30 s, 60 °C × 45 s (except fragment 3, 61 °C × 45 s) and 72 °C × 3 min. Finally, one cycle at 72 °C × 7 min. The ProFlex™ thermocycler 3 × 32-well PCR System (Applied Biosystems™, USA) was used, and the products were visualised on 2% agarose gel.
Sanger sequencing and variant analysisThe PCR products were quantified, diluted to 40 ng/µl and purified with ExoSAP-IT™ for PCR Product Clean-Up (Applied Biosystems, Lithuania). Subsequently, they were sequenced in both directions using the BigDye® Terminator v3.1 Cycle Sequencing kit (Applied Biosystems/Hitachi High Technologies, Japan) and purified with the BigDye® XTerminator™ Purification Kit. Finally, capillary electrophoresis was performed with the 3500 Genetic Analyzer equipment (Applied Biosystems, Lithuania). The consensus sequences were generated using Sequencher 5.4.6 (Gene Codes Corporation, USA).
To confirm the results obtained from the Sanger sequencing, the samples were also analysed at the Sequencing Unit of the University of Antioquia, with support from the PECET (Programa de Estudio y Control de Enfermedades Tropicales [Programme for the Study and Control of Tropical Diseases)]-Immunovirology groups of the same university, by means of NGS using the iSeq™ 100 (Illumina, USA), MiSeq™ (Illumina, USA) and Oxford Nanopore MinION (Oxford Nanopore Technologies, United Kingdom) systems. Variant nomenclature was performed according to the Nextstrain–Nextclade Web 1.10.0 online program, Nextclade CLI 1.7.0 (2021-12-09) and PANGO Lineages (https://cov-lineages.org/).
Statistical analysesThe mutation data were tabulated and analysed using the GraphPad Prism v7.04 program. The concordance of the determination of variants between Sanger sequencing and by NGS was calculated using the kappa index, considering NGS as the gold standard and defining two possible categories (present or absent) in relation to the mutations or changes evidenced in the S protein.
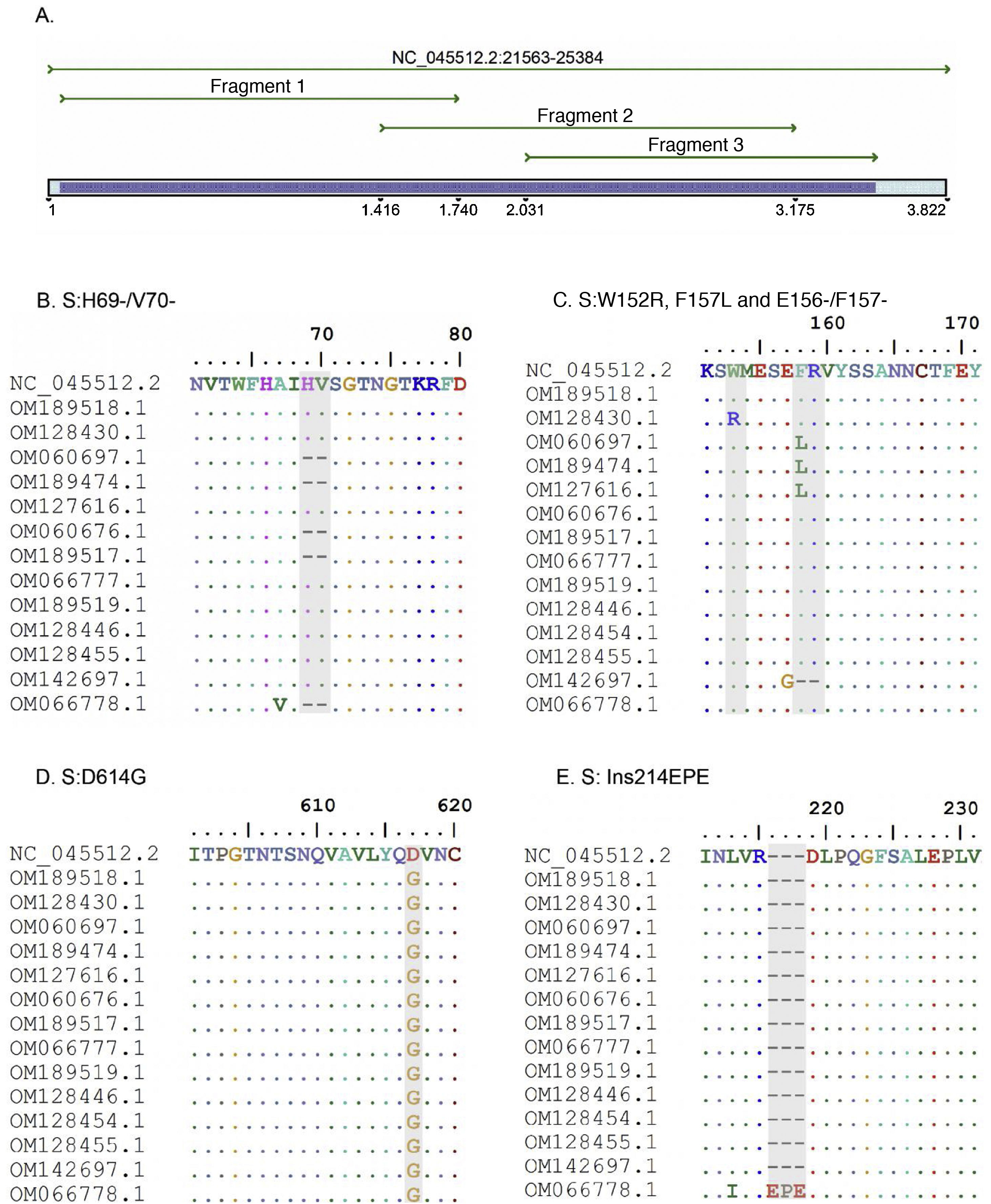
ResultsThree pairs of primers were designed that start at position 21,620 and end at position 25,076 of the reference sequence NC_045512 of the S gene of the SARS-CoV-2 virus, which are listed in Table 1. The amplicons generated overlapped each other and generated a product of 3434 bp with a theoretical coverage of 89.9% of the S gene (3822 bp), as can also be seen in Fig. 1.
List of designed primers.
| S gene | Name of primers | Sequence | Position | Temp (°C) | Amplicon size (bases) |
|---|---|---|---|---|---|
| Fragment 1 | LIMEF1 FW | ACCAGAACTCAATTACCCCCTG | 21620–21641 | 62.5 | 1683 |
| LIMEF1 RV | CTGTGGATCACGGACAGCAT | 23302–23283 | 62.1 | ||
| Fragment 2 | LIMEF2 FW | CTATCAGGCCGGTAGCACAC | 22978–22997 | 62.4 | 1760 |
| LIMEF2 RV | CATGAGGTGCTGACTGAGGG | 24737–24718 | 63.8 | ||
| Fragment 3 | LIMEF3 FW | GACTAATTCTCCTCGGCGGG | 23593–23612 | 63.6 | 1484 |
| LIMEF3 RV | TGCCAGAGATGTCACCTAAATCA | 25076–25054 | 60.1 |
Temp: melting temperature.
.")
A. In silico simulation of coverage of the S gene by the LIME primers. The products generated cover most of the mutations and deletions associated with VOI and VOC. B. H69/V70 deletion common in the alpha variant. C. Point mutations and deletions among amino acids 148, 150 and 160 of the S protein; particularly noteworthy is the E156/F157 deletion identified in the delta variant. D. Mutation D614G present in all the samples evaluated. E. 214EPE insertion characteristic of the omicron variant 21K (BA.1).
According to the in silico analysis, the mutations present in the variants reported to date do not interfere with the yield and amplification of the fragments. However, a single nucleotide change was identified in the LIMEF3 FW primer that amplifies fragment 3.
In silico analysis of variants of interest and concernIn silico analysis of the VOI and VOC reported to date in the World Health Organization’s Tracking SARS-CoV-2 variants was conducted. Theoretically, with the Sanger methodology, the specific mutations S:G75V, S:T76I, S:F490S and S:T859N, and seven deletions of the 21G variant (lambda) corresponding to the C.37 lineage can be detected. In the case of the mu variant, the relevant S:R346K and S:Y144S mutations can be detected, as well as the seven mutations also reported in the other VOI and VOC.
For the VOC, in alpha, the Sanger results identified the specific mutations S:A570D, S:T716I and S:S982A. However, the mutation located at amino acid 1118 (S:D1118H) could not be detected. In the case of the beta variant, mutations S:D80A and S:D215G and deletions S:L241-, S:L242- and S:A243- can be detected, and in gamma, variants S:D138Y, S:R190S and S:T1027I can be detected. For the delta variant, the mutations shared by clade 21A and 21J (S:R158G, E156- and S:F157-) could be detected, but subclade 21A could not be differentiated from 21J due to the presence of mutations in the genes of the nucleocapsid and ORF1a-b, which are not analysed by the Sanger methodology. Finally, in the case of the omicron variant, two mutational profiles close to each other were found, clades 21K and 21L. For the 21K clade, eight specific mutations could be detected, but in the 21L clade only seven mutations would be detected, which would allow differentiation between the two clades.
Analysis of variants in positive samples for SARS-CoV-2The amplicons obtained from the three conventional PCRs experimentally cover 3434 nucleotides of the S gene, corresponding to 1144 amino acids. Good quality Sanger sequences were obtained in all 15 clinical samples. Two alpha samples, three gamma, one delta, three mu and one omicron sample were classified and five strains were related to the initial isolate of the Wuhan-Hu-1 virus.
In the blinded comparison of the results of the Sanger sequencing and NGS (gold standard), the classification results of both methods in the 15 samples were concordant (Appendix B Supplementary material, annex 1).
Samples B1 (OM189518) and 5703 (OM128430) were classified in clade 19B, while the NGS result for both samples placed them in 20A. The mutations found were S:D614G for sample B1, and the mutations W152R and D614G were found in 5703. For their part, the sequences with codes 6041 (OM060697), 2403 (OM189474) and 12,422 (OM127616) were diagnosed in early 2021 and classified with pangolineage B.1.625, clade 20A.
Regarding the identification of VOC, samples 11072 (OM060676) and BC08 (OM189517) were located in clade 20I (alpha, V1). The constant mutations detected in these samples were 69del, 70del, 144del, N501Y, A570D, D614G, P681H, T716I and S982A. Similarly, three samples were classified as 20J (gamma, V3), corresponding to codes 11,896 (OM066777), BC22 (OM189519) and 28,657 (OM128446). The main mutations detected were D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y and T1027I.
Sample 36,211 (OM142697) was classified by Sanger as a delta variant, but was located in clade 21A, while it was located in 21J by the NGS methodology. Both sequencing methods present the same S protein mutation profile. Sample M1 (OM066778) was categorised as an omicron variant in the 21K clade, with more than 36 mutations detected in the S protein amino acid sequence.
VOI identification was also achieved. Samples 30,535 (OM128454), 12,932 (OM128455), and BC13 (OM301638) were classified as mu and located in clade 21H. Common mutations detected were S:T95I, S:Y145N, S:R346K, S:E484K, S:N501Y, S:D614G, S:P681H and S:D950N.
DiscussionThe comparison of the results of the Sanger and NGS sequencing of the COVID-19 samples evaluated showed that the Sanger methodology achieves the correct classification of different lineages of SARS-CoV-2 with results consistent with those obtained by NGS (Supplementary material, annex 1).
The statistical analysis with the kappa index was 0.146 (95% CI −0.032 to 0.325), showing a slight agreement between methods, probably associated with the fact that none of the methodologies detect all the mutations reported in the Tracking SARS-CoV-2 variants database. It should be noted that the Sanger methodology detects key mutations to identify the clade to which a strain belongs.
Although technically it is not possible to amplify and sequence the 3822 bases of the S gene using the Sanger methodology in a single reaction13, in our study we proposed the amplification of three overlapping fragments to achieve 90% coverage of the S gene. This is efficient compared to other sequencing methodologies13,14, since it shortens processing time, does not require a minimum number of samples for analysis, reduces the number of reactions necessary to obtain results and reduces the associated costs.
The methodology proposed in this article requires adequate collection, conservation and transport of the samples, which ensures a complete RNA with cycle threshold values less than 25. These data are similar to those recommended in other investigations13,15. The protocol was tested with samples extracted by automated methods of magnetic beads and extractions with silica columns, generating good results, particularly with the column methodology.
Although not all the SARS-CoV-2 variants were found in the clinical samples, the in silico analysis of protein S showed that this methodology guarantees the detection of the amino acids of interest S:K417, S:L452, S:S477, S:T478, S:E484, S:F490, S:S494 and S:N501. These mutations are of great importance because the WHO and the United States’ CDC defined them as key mutations present in VOI and VOC for genomic surveillance purposes13.
The S:D614G mutation was found in all the samples analysed. It is called the universal mutation and all VOI and VOC are considered to be derived from it16. S:D614G is related to increased viral replication in the upper respiratory tract and increases the infection’s efficiency15. This adaptive advantage is related to the increase in COVID-19 cases in some European countries in mid-2020 and the subsequent worldwide spread17,18.
The alpha variant is associated with multiple mutations in the S protein, the most prominent one being S:N501Y and the 69–70 deletion. The latter generates a failure of some real-time PCR assays5,19 and is found in other variants, such as 20A, 21K (omicron) and 21L (omicron).
Based on covariate data, mutations S:A570D, S:T716I, S:S982A and S:D1118H are associated with the alpha variant16. In this study, the S:D1118H mutation could not be detected due to lack of coverage in that region. However, the mutations identified are sufficient to categorise the variant.
Gamma is associated with multiple mutations; the specific ones are S:N501Y, S:E484K, S:L18F, S:K417T and S:H655Y. They are shared by several VOC, except the delta variant16. Our methodology is able to detect around eight of the characteristic mutations, including three specific mutations: S:D138Y, S:R190S and S:T1027I.
The delta variant and the omicron variant are considered to be VOC, and their rapid spread has been the cause of the outbreaks since mid-202120. The delta variant has two subclades; the Sanger and NGS results analysed in Nextstrain generated the classification 21A and 21J, respectively, for sample 36211. The discrepancy obtained by the Sanger and NGS methods is due to the fact that the 21J subclade contains all the mutations of the 20A clade with additional mutations in the N gene (N:G215C), as well as in ORF1a (ORF1a:A1306S, ORF1a:V2930L, ORF1a:T3255I, ORF1a:T3646A) and ORF1b (ORF1b:A1918V, and ORF7b:T40 I)16,21, which are not analysed in this study.
The omicron variant emerged in November 2021 and early infections and sequence data are predominantly from South Africa22. The identification of this variant is very significant because it is currently the most common one, its mutations are associated with greater transmissibility, immune evasion and greater binding affinity to ACE223,24. It shares two mutations with the alpha VOC but has around 26 specific mutations16, which are mostly detected by the Sanger method.
The mu VOI emerged in early 2021 and was very common in Colombia16. This variant has mutations that are also found in VOC and VOI such as S:T95I, S:E484K, S:N501Y, S:P681H, and S:D95N. The S:R346K mutation has been considered specific for the mu variant, although this mutation also occurs in the omicron variant.
It was not possible to obtain samples from the beta and lambda variants to perform the experimental analysis; the in silico analyses confirm that the Sanger sequencing method can detect characteristic mutations.
Our group acknowledges that the number of samples analysed is low, but it allows us to demonstrate that the methodology has the capacity to experimentally categorise VOC and VOI using freely accessible online applications without requiring the creation of subjective algorithms. For further studies, we recommend a larger number of samples and other variants not included in the study.
This methodology overcomes some of the limitations of25, intercalating26 and allele-specific real-time probe PCR methods27, where the use of specific primers and/or probes is necessary to evaluate each mutation and with prior knowledge of circulating mutations. Additionally, some of these PCR are not specific for determining genetically related variants (subclades), they do not allow the appearance of new variants to be evaluated and they underestimate the real number of circulating variants.
Recently, Sanger sequencing methodologies that study the S protein have been published and even standardised commercial protocols are already available28–30. Several of those studies require a larger number of primers targeting domains of interest, such as receptor-binding domains and receptor-binding motifs30, as well as analysing mutations in other regions28, and in some cases they perform a total sequencing of the S protein. All these previous methodologies involve performing multiple PCRs29, which increases the cost compared to the methodology that we propose here.
It is important to stress that this approach does not replace NGS technology and other PCR-based assays. Rather, it seeks to increase the capacity of the epidemiological surveillance network, being used as a variant screening method, and in cases in which NGS has little coverage or depth it can be used as a strategy to complete the sequences.
In conclusion, this methodology enables the detection of mutations in the S protein in a streamlined and precise way, quickly and with infrastructure that is available in clinical, public health and research laboratories in Colombia that serve as support for the genomic diagnostic and surveillance network of the SARS-CoV-2 virus.
FundingThis work was financed with resources from Colombia’s general awards system (BPIN 2020000100152) and from the University of Antioquia.
Conflicts of interestThe authors declare that they have no conflicts of interest.
The following is Supplementary data to this article:
