


El síndrome de insensibilidad parcial a andrógenos (PAIS) es un trastorno de la diferenciación sexual 46 XY en el que existe una pérdida de función del receptor de andrógenos (RA). Se trata de una enfermedad causada por mutaciones en el gen AR (Xq11-12) que codifica para el RA.
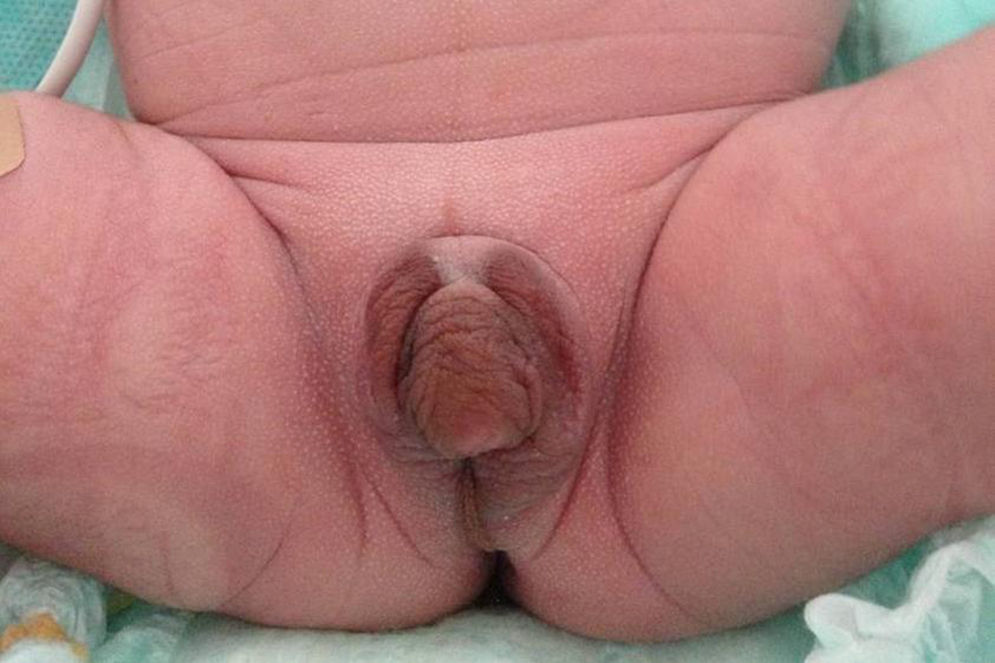
Presentamos el caso de un recién nacido a término producto de un embarazo controlado sin complicaciones, segundo hijo de padre de origen italiano y madre dominicana, sanos y no consanguíneos. Ecografías prenatales normales con diagnóstico de sexo femenino. Peso al nacimiento 2.862g (−0,12DE), longitud 46,5cm (−0,62DE), perímetro cefálico 35cm (1,16DE), con Apgar 8-9. Presentaba genitales ambiguos, hiperpigmentados, con un tubérculo genital de 1,5cm con meato urinario hipospádico, rodetes de labios escrotales simétricos, fusionados en su porción posterior, sin abertura vaginal ni testes a la palpación, correspondiente a Prader 4-5 (fig. 1). Presentó un distrés respiratorio leve, secundario a una hipertensión pulmonar con una miocardiopatía hipertrófica no obstructiva, que se resolvió en 2 semanas. El equilibrio ácido-base y los niveles de sodio y potasio fueron normales. La ecografía abdominal realizada el primer día de vida demostró una estructura compatible con útero y una cavidad endometrial rellena de líquido, no visualizándose ovarios ni testes.

El estudio hormonal en la primera semana de vida mostró LH<0,1 UI/ml, FSH 0,4mUI/ml, testosterona 152ng/dl, cortisol 7μg/ml, ACTH 23,3pg/ml, androstendiona 5,5ng/ml, DHEAS 159μg/ml, aldosterona 1.428pg/ml, 17-hidroxiprogesterona 9ng/ml, renina 12,2ng/ml/h y estradiol 17-beta<12pg/ml, dihidrotestosterona 28,2ng/dl, todos dentro de valores normales. Posteriormente se solicitó: 11-desoxicortisol 6,15 ng/ml, desoxicorticosterona 3ng/ml, inhibina B 99pg/ml y hormona antimülleriana 18,8ng/ml, también en valores normales para la edad.
El cariotipo fue 46 XY, compatible con trastorno de la diferenciación sexual 46 XY con genitales ambiguos y el estudio de la microdeleción del gen SRY resultó normal. Se solicitó estudio del gen del receptor de los andrógenos y del gen SRD5A2, asociado al déficit de 5-alfa-reductasa, ya que la madre nació en la comarca de la República Dominicana donde se describió este síndrome.
La cistoureterografía miccional seriada evidenció una uretra de morfología masculina con un seno urogenital y un nuevo control ecográfico a los 10 días de vida reveló la presencia de estructuras paravesicales compatibles con gónadas sin folículos en su interior, de 0,8 y 1cm. Ante el hallazgo del sexo genético 46 XY, la producción de testosterona en niveles normales por una estructura ajena a la suprarrenal (en un probable tejido testicular posiblemente en la gónada), uretra de longitud y trayecto de características masculinas, y la existencia de un pene de tamaño aceptable y tras el acuerdo multidisciplinar entre endocrinología infantil, cirugía infantil, neonatología y la familia, a los 18 días de vida se le asignó sexo masculino.
A los 36 días de vida fue sometido a cirugía por una hernia inguino-escrotal izquierda, realizándose orquidopexia y biopsia gonadal. La biopsia testicular evidenció hipoplasia germinal marcada (tipo II de Nistal et al.1) y cariotipo gonadal 46 XY.
Ante el hallazgo de un recién nacido con genitales ambiguos y una estructura compatible con útero en ecografía de urgencia, el diagnóstico más probable es una virilización de una mujer 46 XX, siendo la causa más frecuente la hiperplasia suprarrenal congénita por el déficit de 21-hidroxilasa. Al demostrase un sexo genético 46 XY, el trastorno de la diferenciación sexual 46 XY es más complejo de filiar2. El estudio del gen SRY, principal gen implicado en la diferenciación gonadal primaria fue normal. Los restos de vagina y útero podrían corresponder con un síndrome de persistencia de los conductos müllerianos, por déficit en la producción de la hormona antimülleriana o por déficit en el receptor. La infravirilización podría deberse a su vez a un PAIS, aunque no suele asociar restos müllerianos. Un déficit de 5-alfa-reductasa podría explicar parte del cuadro, y la madre procede de Las Salinas3, la zona de la República Dominicana donde se descubrió esta enfermedad, pero donde además existe un muy elevado porcentaje de quimera ovotesticular y de disgenesia gonadal mixta. Otros síndromes asociados, que conllevan nefropatía, alteraciones cerebrales o hipertrofia cardiaca, como el síndrome de LEOPARD, son poco probables, pero hay que evaluarlos según los hallazgos.
A los 2 meses se dispuso del estudio molecular del gen AR, que detectó la mutación c.1139C >G que sustituye la prolina en posición 380 por arginina, asociada al PAIS, lo que confirmó el diagnóstico. Esta mutación se ha descrito únicamente en otro paciente (Audi et al., 2010)4 procedente de Gambia, con PAIS y restos müllerianos. El estudio del gen SRD5A2 demostró un cambio en homocigosis en la posición 265 (c.265C >G), con sustitución de leucina por valina (p.Leu89Val), de significado incierto5.
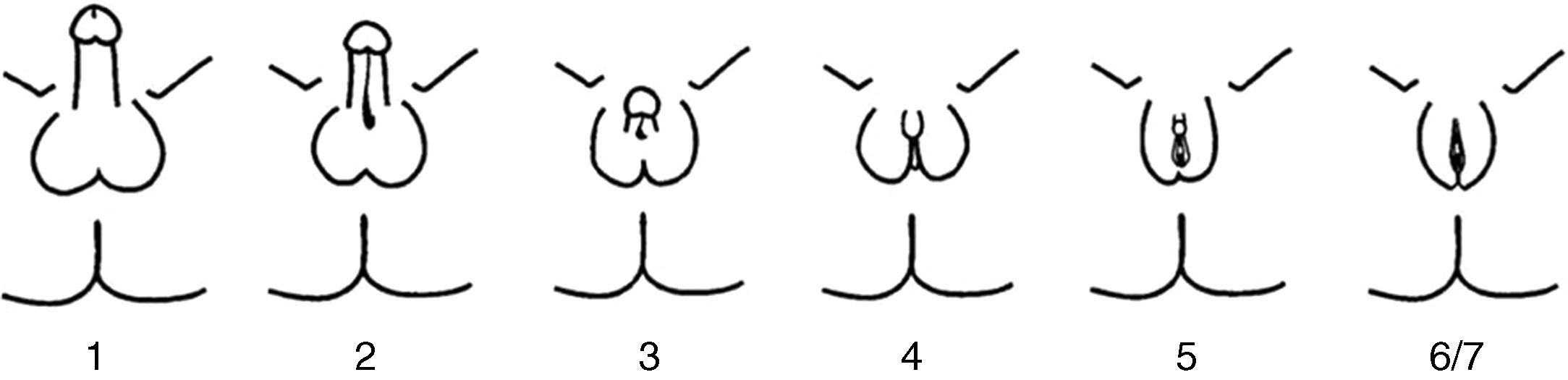
El PAIS es una enfermedad recesiva ligada al cromosoma X, causada por mutaciones en el gen AR. Este gen está situado en la región Xq11-12 y codifica para el RA6. Tan solo se encuentra una mutación AR en un 20% de los pacientes con PAIS. La prevalencia del PAIS es desconocida debido a la variabilidad de las manifestaciones clínicas y la existencia de formas sutiles o atípicas. Se estima que puede ser 10 veces menos frecuente que en el síndrome de insensibilidad total a andrógenos (CAIS). Se presenta en individuos 46 XY con un espectro clínico muy variable, desde la feminización casi completa hasta la masculinización casi normal, pasando por individuos con ambigüedad sexual manifiesta. La variabilidad clínica es reflejo de los distintos tipos de mutaciones que pueden afectar al gen del RA. El fenotipo predominantemente masculino exhibe micropene, hipospadias variable y criptorquidia, durante la pubertad presentan aspecto eunucoide y desarrollo mamario (ginecomastia). El fenotipo predominantemente femenino se manifiesta con clitoromegalia, fusión de los labios y vello púbico durante la pubertad. Las formas más ambiguas se descubren al nacimiento o durante la infancia. La clasificación fenotípica correcta no es la de Prader, sino la de Quigley et al.7 (fig. 2). Los genitales internos, derivados del conducto de Wolff pueden estar parcial o totalmente desarrollados mientras que los testículos se encuentran con mayor frecuencia no descendidos (en región inguinal o en escroto/labios mayores), carecen o tienen escasa dotación de células germinales y son más susceptibles de malignización en el adulto. Aunque se refiere que no deben existir restos mullerianos, existen casos puntuales descritos de persistencia de estas estructuras en pacientes con CAIS8–10 o PAIS, como en nuestro paciente; hasta el presente no están aún claramente dilucidado el mecanismo de esta asociación. El pronóstico depende del grado de ambigüedad de los genitales y de la adecuación física y psicosocial al sexo asignado. Pueden presentar complicaciones en la edad adulta con mayor tendencia a la malignización de las gónadas, infertilidad y osteoporosis.

Cuando existe ambigüedad genital, la asignación de sexo es un proceso urgente pero complejo, que requiere la evaluación por un equipo multidisciplinar que abarca la familia, neonatólogos, radiólogos, cirujanos pediátricos, endocrinos pediátricos y psicólogos. Debe ser resuelta lo más pronto posible, pero solamente cuando se disponga de información suficiente, pensando en el mejor interés del recién nacido11.