


Cambios metabólicos in utero establecen patrones fisiológicos y estructurales a largo plazo que pueden «programar» la salud durante la vida adulta, teoría popularmente conocida como «Hipótesis de Barker». Evidencia experimental y clínica sugiere que patologías como hipertensión arterial, enfermedad isquémica coronaria, síndrome metabólico y diabetes mellitus tipo 2, pueden «programarse» durante las primeras etapas del desarrollo fetal y manifestarse en etapas tardías, al interactuar con el estilo de vida y otros factores de riesgo adquiridos convencionales con el medio ambiente. El objetivo de esta revisión es presentar evidencia adicional y actualizada que apoyen la asociación entre la salud fetal intrauterina y el aumento en la prevalencia de enfermedades crónicas no transmisibles en la edad adulta. La función endotelial, el estrés oxidativo, la resistencia a la insulina, y la función mitocondrial son tratadas como posibles mecanismos celulares y moleculares.
Adverse events during intrauterine life may program organ growth and favor disease later in life. This is the usually called ‘Barker's hypothesis’. Increasing evidence suggests that conditions like vascular disease, hypertension, metabolic syndrome, and type 2 diabetes mellitus are programmed during the early stages of fetal development and become manifest in late stages of life, when there is an added impact of lifestyle and other conventional acquired environmental risk factors that interact with genetic factors. The aim of this review was to provide additional, updated evidence to support the association between intrauterine fetal health and increased prevalence of chronic non-communicable diseases in adulthood. Various potential cellular and molecular mechanisms proposed to be related to the above hypothesis are discussed, including endothelial function, oxidative stress, insulin resistance, and mitochondrial function.
En las últimas décadas diversas áreas de investigación han sugerido que los eventos implicados en el desarrollo fetal normal tienen efectos a largo plazo e influyen en la salud durante la vida adulta1–7. Se piensa que los cambios metabólicos in utero pueden establecen patrones fisiológicos y estructurales a largo plazo, que «programan» la salud durante la vida adulta2–4. Los estudios de Barker et al.5–7 en la década de 1980, establecieron que la prevalencia de algunas enfermedades en el adulto, como ateroesclerosis, hipertensión arterial (HTA), accidente cerebrovascular, diabetes mellitus tipo 2 y dislipidemias, se relacionaban con el ambiente intrauterino («Hipótesis de Barker»). Actualmente, esta hipótesis se conoce como el origen durante el desarrollo de la salud y enfermedad6,7.
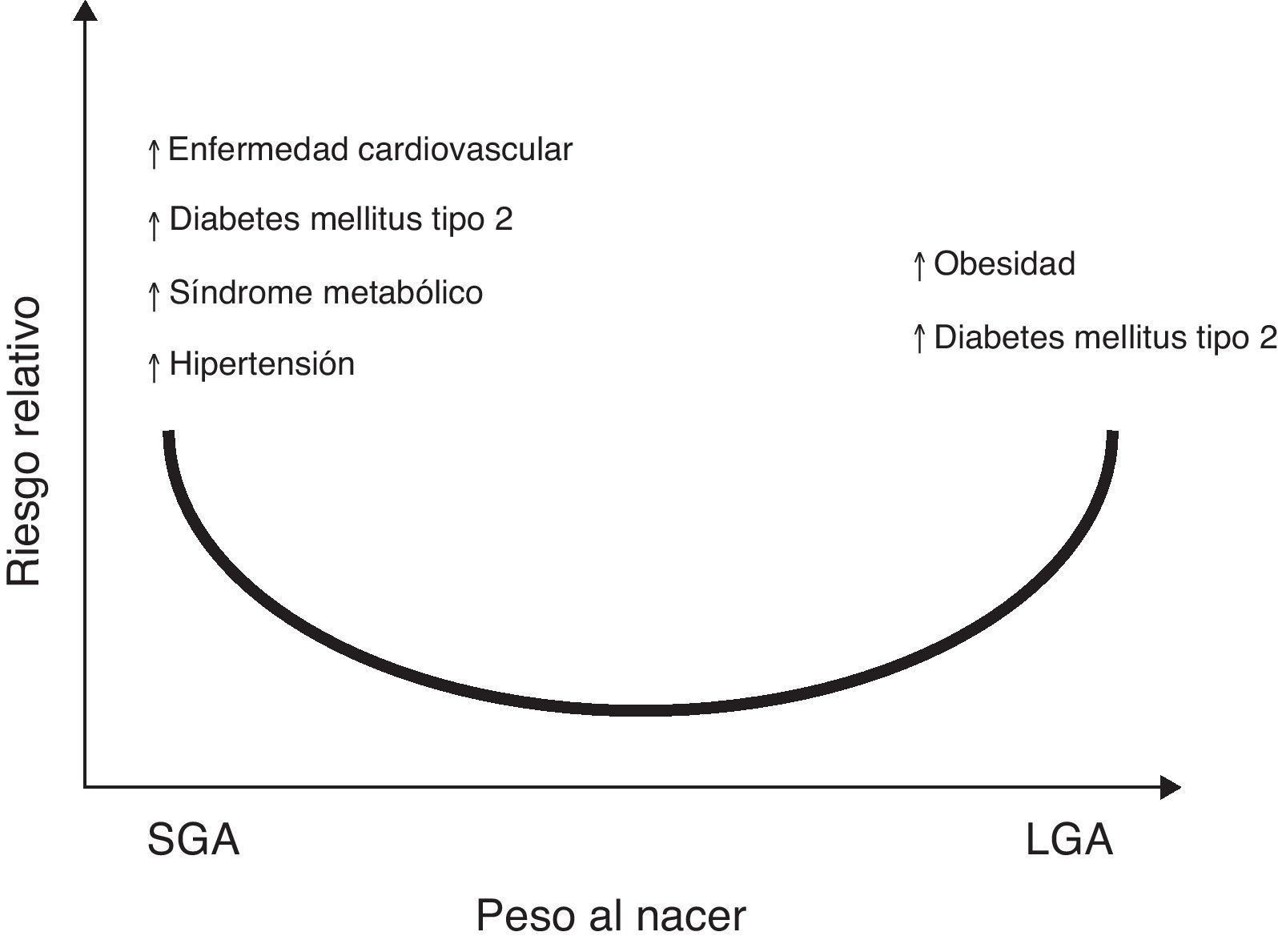
Aunado a lo anterior, se ha reportado la asociación entre el bajo peso y la talla al nacer, con el aumento en el riesgo de sufrir posteriormente enfermedades como hipertensión arterial (HTA), síndrome metabólico y accidente cerebrovascular7,8. Cambios en el peso o en la composición corporal al nacer, ya sea en el rango superior de la normalidad para la edad gestacional (LGA por sus siglas en inglés, large for gestational age), o reducciones significativas en el tamaño y peso al nacer, (SGA por sus siglas en inglés, small for gestational age), podrían dar lugar a secuelas metabólicas en la etapa adulta9,10 (fig. 1).
 incrementa el riesgo de obesidad y síndrome metabólico en la etapa posnatal121. De la misma manera, presentar un bajo peso para la edad gestacional (SGA: small for gestational age), indicaría mayor riesgo para presentar diabetes mellitus tipo 2, enfermedad cardiovascular, síndrome metabólico, hipertensión arterial y obesidad en la vida adulta122.")
Riesgo relativo entre el peso al nacer con la prevalencia de enfermedades crónicas no transmisibles en la vida adulta. Sobre la base de observaciones epidemiológicas y experimentales y la «hipótesis de Barker»5–7, existe suficiente evidencia que apoya que un aumento en el peso para la edad gestacional (LGA: large for gestational age) incrementa el riesgo de obesidad y síndrome metabólico en la etapa posnatal121. De la misma manera, presentar un bajo peso para la edad gestacional (SGA: small for gestational age), indicaría mayor riesgo para presentar diabetes mellitus tipo 2, enfermedad cardiovascular, síndrome metabólico, hipertensión arterial y obesidad en la vida adulta122.
Posteriormente, otras investigaciones confirmaron esta relación y en la actualidad se tratan de esclarecer algunos de los mecanismos11,12. A nivel experimental, la restricción nutricional durante la gestación ha mostrado que afecta irreversiblemente la estructura, el metabolismo y la función en algunos órganos «programando» su descendencia a patologías futuras13,14.
Mecanismos relacionados con la Programación FetalLa hipótesis sobre los orígenes relacionada con la Programación Fetal propone que la enfermedad coronaria, el accidente cerebrovascular, la HTA y la diabetes mellitus tipo 2, se originan en la plasticidad del desarrollo que ocurre en respuesta a factores maternos y placentarios durante la vida fetal y la lactancia. Por ejemplo, se ha postulado que la HTA puede ser causada por el menor número de glomérulos que tienen las personas de talla pequeña como infantes con bajo peso al nacer (BPN) y con retardo en el crecimiento intrauterino (RCIU)15,16.
El segundo proceso comprende la regulación hormonal y metabólica. Un recién nacido prematuro o con BPN, presenta mayor susceptibilidad de tener un patrón metabólico «ahorrativo» o «thrifty phenotype hypothesis» para el manejo de los nutrientes5–7. La resistencia a la insulina y un incrementado estado de estrés oxidativo (EO), que se asocia al BPN, podrían ser considerados desde esta perspectiva, como la persistencia de la respuesta fetal para la preservación de glucosa en el cerebro, a expensas del transporte de este carbohidrato al músculo, para su propio metabolismo y crecimiento. Según la hipótesis del fenotipo «ahorrador» un crecimiento fetal pobre, ocasionaría una disminución en el número de células pancreáticas β y una disminución de la capacidad de producir insulina, conduciendo en la edad adulta a estados de resistencia a la insulina17. La evidencia de que recién nacidos con BPN o con RCIU presentarán resistencia insulínica es consistente5–7. En una revisión sistemática publicada en 2008 por Whincup et al.18 se reportó que, en la mayoría de las poblaciones estudiadas, el peso al nacimiento se hallaba inversamente relacionado con el riesgo de padecer diabetes mellitus tipo 2 e HTA.
Una tercera asociación entre el BPN y la Programación Fetal, es que las personas de talla pequeña al nacer son más vulnerables a las influencias ambientales adversas durante las etapas posteriores de la vida19,20. Sin embargo, no existe una clara asociación entre peso elevado y mayor riesgo de HTA21,22.
Alimentación durante la gestación, peso al nacer y programación metabólicaLos experimentos con animales, y las observaciones epidemiológicas en humanos, sugieren que la nutrición recibida en el ambiente intrauterino modula la función de varios tejidos con actividad metabólica en la vida posnatal23,24. Una importante consecuencia de la restricción calórica y de nutrientes es el aparente y acelerado periodo de crecimiento posnatal25. En humanos, se ha observado que la relación entre pobreza y desnutrición durante la gestación, aumenta la morbilidad y mortalidad infantil7. Sobre este último, Barker y su grupo26, evaluaron la tolerancia a la glucosa en individuos de 64 años; sus resultados mostraron una fuerte asociación entre bajo peso al nacimiento y enfermedades metabólicas. Los neonatos con peso menor de 2.500g al nacimiento, tenían 7,5 mayor riesgo de padecer diabetes mellitus tipo 2 o intolerancia a la glucosa, en comparación con los que pesaron más 4.000g.
Al mismo tiempo, se ha observado que el crecimiento acelerado o «catch up» se da aproximadamente en 85% de los niños prematuros o con BPN en los primeros 3 años de vida. Singhal et al.27,28 y Ross et al.29 observaron una asociación entre el crecimiento rápido posnatal temprano y un aumento en el riesgo de presentar indicadores de síndrome metabólico y una composición corporal desfavorable. Una hipótesis similar a la anterior es la del «catch-up growth» propuesta por Arends et al.30 y Cianfaranri et al.31. Esta se basa en que recién nacidos con restricción nutricional, presentan niveles menores de insulina y de factores de crecimiento similares a la insulina como (IGF-1) e IGF unido a la proteína 3 (IGFBP-3). Aunque la normalización de los niveles de estas hormonas generalmente ocurre durante los primeros 3 meses de vida posnatal, lo cual coincide con el rápido crecimiento observado en estos neonatos, es posible que los tejidos que han estado con deficiencia crónica de insulina y de IGF-1, y que de manera súbita deban enfrentarse a altas concentraciones de estas hormonas, desarrollen resistencia a la insulina como mecanismo de defensa hacia la hipoglicemia. De esta manera se explicarían las asociaciones encontradas entre el «catch-up growth» y el aumento en el riesgo de síndrome metabólico y obesidad posnatal29–33.
Además de la composición corporal alterada en la restricción nutricional materna, un incremento en el contenido de triglicéridos intramusculares (IMTG) ha sido encontrado en hijos con alteraciones en el crecimiento intrauterino, que puede en parte, explicar su predisposición a diabetes mellitus tipo 232. Cambios en los niveles de IMTG se asocian con la reducción de la actividad de la enzima carnitina palmitoil-1 (CPT-1b), una proteína clave en la oxidación de estos ácidos grasos33.
Asimismo, una buena placentación y un adecuado desarrollo fetal dependen también de la concentración de otras importantes hormonas como leptina34 y adiponectina placentarias35. En la vida posnatal estas hormonas son secretadas principalmente por el tejido adiposo y participan en la regulación del metabolismo, de la función cardiovascular y de la homeostasis energética entre otros. En el adulto, la leptina regula el consumo y gasto energético a largo plazo, mientras que la adiponectina tiene propiedades antiinflamatorias e incrementa la sensibilidad a la insulina36. Se ha observado un aumento en las concentraciones de leptina en fetos y placentas de madres diabéticas y una disminución en los niveles de adiponectina en sus hijos al nacimiento. En neonatos con RCIU, los niveles de estas 2 adipoquinas están disminuidos37. Sin embargo, se ha observado que al año de edad estos niños presentan niveles aumentados de leptina al compararlos con niños con peso al nacer adecuado, y en adultos con antecedente de BPN se han observado niveles aumentados de esta hormona al comparar con sujetos con igual índice de masa corporal38. En ratas sometidas a desnutrición in utero, el aumento de leptina en la etapa posnatal temprana ha sido asociado a obesidad e indicadores de síndrome metabólico en la edad adulta39. Además, el peso al nacer en los valores extremos bajo y alto se ha relacionado con un mayor porcentaje de grasa en etapas posteriores de la vida40. Posiblemente el nivel de adipoquinas en etapas tempranas del desarrollo juega un papel importante en la programación de la composición corporal del individuo. En humanos, se han observado estados de hiperleptinemia en obesidad, síndrome metabólico y enfermedad cardiovascular en sujetos con BPN41.
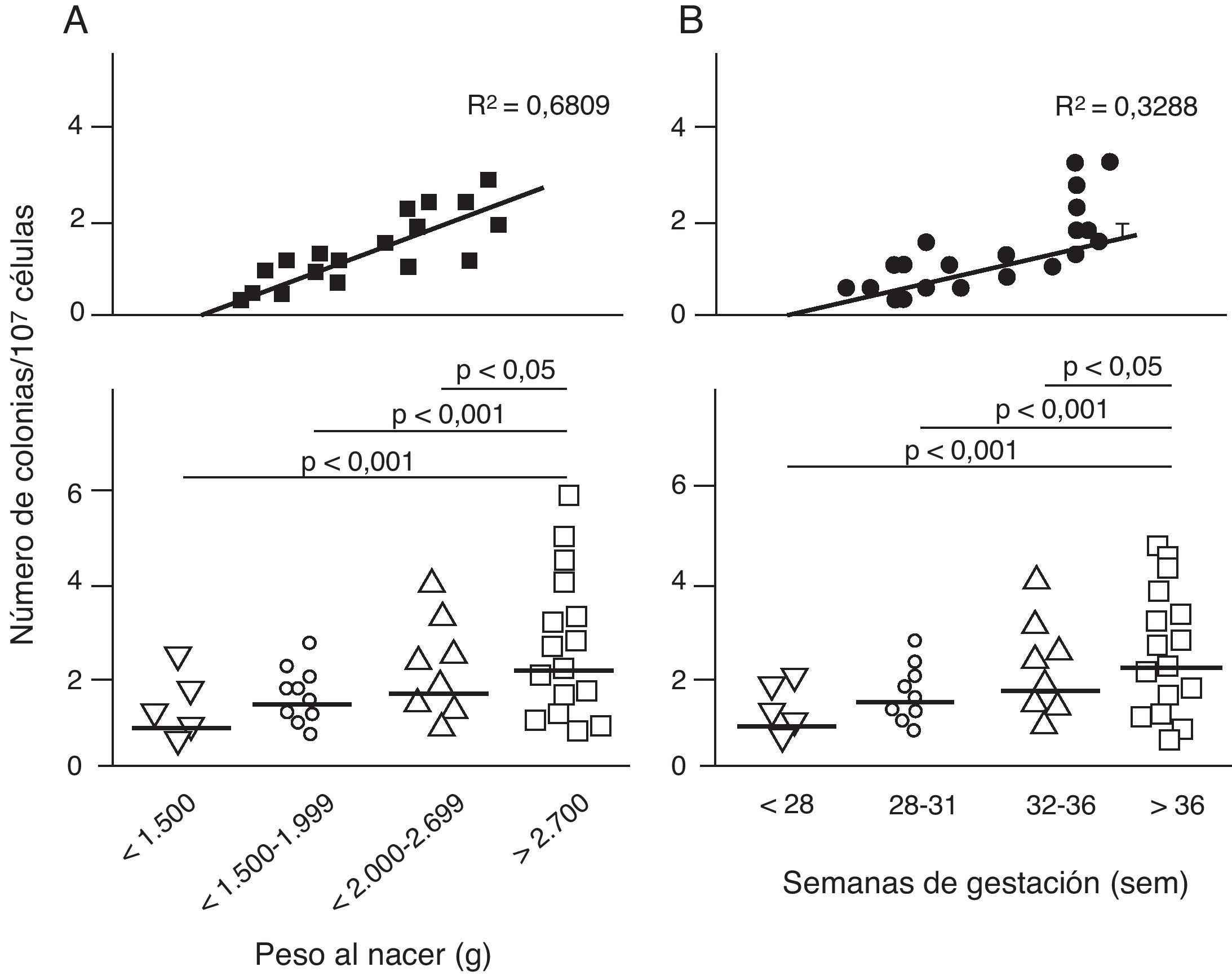
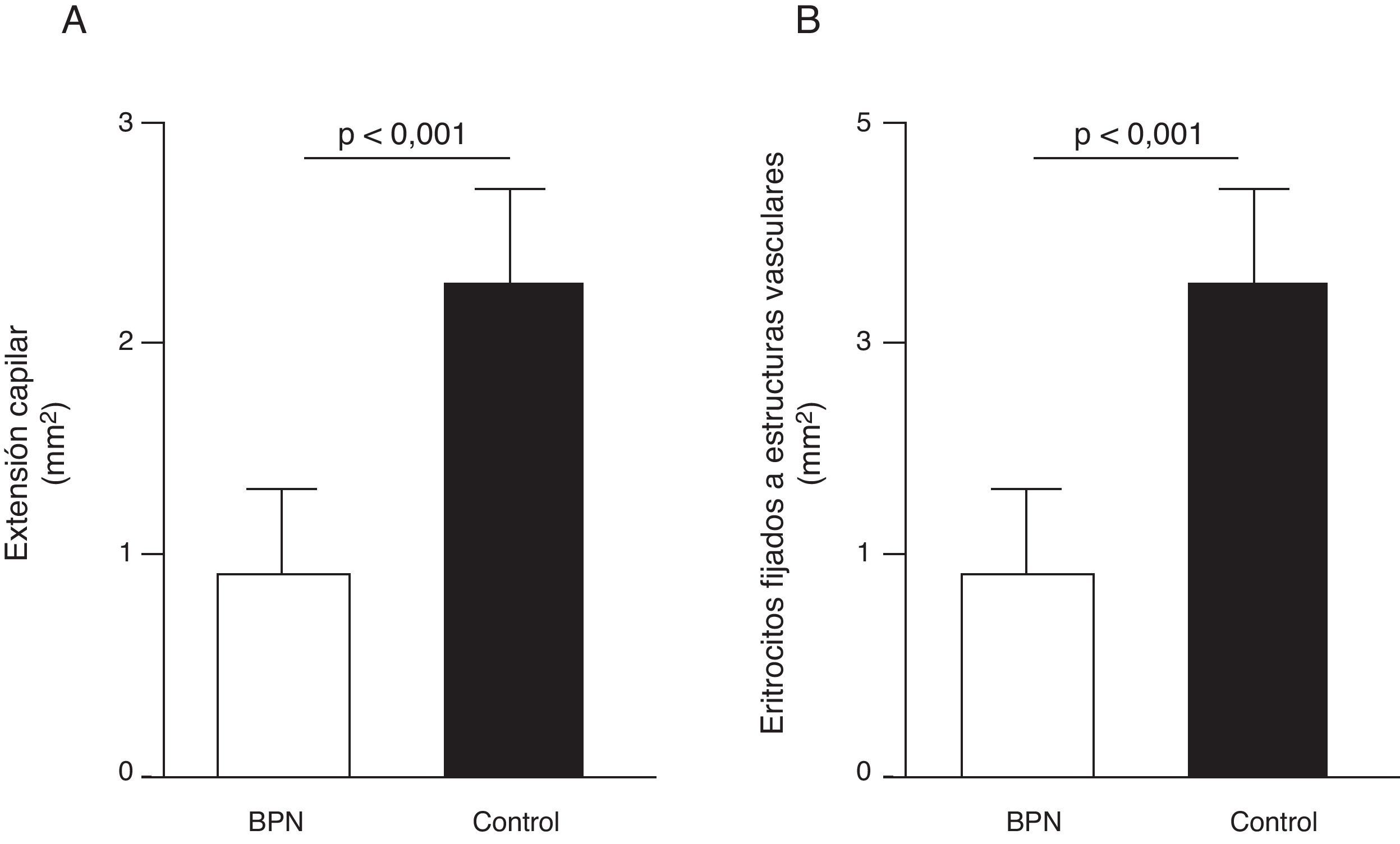
Disfunción endotelial e hipertensión del adultoLos mecanismos que median la Programación Fetal de la HTA del adulto son numerosos42–44. Por ejemplo, varios autores han demostrado relación con cambios en la función renal (reducción del número de nefronas), el sistema neuroendocrino (desregulación del eje hipotálamo-pituitaria- adrenal), y el sistema vascular (disfunción vascular y reducción de la densidad de las arteriolas y capilares)45,46. Recientemente, Ligi et al.47 centraron su hipótesis en el origen de las anomalías vasculares y las propiedades angiogénicas endoteliales de las células formadoras de colonias (ECFC) presentes en la sangre del cordón umbilical de infantes con BPN, en comparación con bebés nacidos a término y mediante parto normal. Las ECFC son un tipo de células que se caracterizan por su capacidad de formar colonias de células endoteliales in vitro y reparar los daños ocasionados por el cambio en el fenotipo vascular (fig. 2A y B). Así, Ligi et al.47 reportaron que colonias cultivadas de ECFC provenientes de niños con BPN, presentaban una reducida capacidad para formar estructuras tubulares y capilares, menor capacidad proliferativa y menor potencial angiogénico, (fig. 3A y B).
 en las propiedades angiogénicas endoteliales de células formadoras de colonias (ECFC) presentes en la sangre del cordón umbilical. Puede observarse que las células cultivadas de ECFC provenientes de infantes con BPN, presentaron menor capacidad de formar colonias (fig. 2A). Este hallazgo también fue encontrado en relación con el tiempo gestacional y el peso al nacer (fig. 2B). Tomado y modificado de Ligi I, et al. Blood. 2011;118:1699-709.")
Efecto del bajo peso al nacer (BPN) en las propiedades angiogénicas endoteliales de células formadoras de colonias (ECFC) presentes en la sangre del cordón umbilical. Puede observarse que las células cultivadas de ECFC provenientes de infantes con BPN, presentaron menor capacidad de formar colonias (fig. 2A). Este hallazgo también fue encontrado en relación con el tiempo gestacional y el peso al nacer (fig. 2B). Tomado y modificado de Ligi I, et al. Blood. 2011;118:1699-709.
 en las propiedades proliferativas endoteliales de células formadoras de colonias (ECFC) presentes en la sangre del cordón umbilical. Puede observarse que colonias cultivadas de ECFC provenientes de infantes con BPN, presentaron reducida capacidad para formar estructuras tubulares y capilares (fig. 3A), menor capacidad proliferativa y menor potencial angiogénico (fig. 3B). Tomado y modificado de Ligi I, et al. Blood. 2011;118:1699-709.")
Efecto del bajo peso al nacer (BPN) en las propiedades proliferativas endoteliales de células formadoras de colonias (ECFC) presentes en la sangre del cordón umbilical. Puede observarse que colonias cultivadas de ECFC provenientes de infantes con BPN, presentaron reducida capacidad para formar estructuras tubulares y capilares (fig. 3A), menor capacidad proliferativa y menor potencial angiogénico (fig. 3B). Tomado y modificado de Ligi I, et al. Blood. 2011;118:1699-709.
Los anteriores hallazgos proporcionan la primera evidencia en humanos de la relación entre el peso al nacer y las propiedades angiogénicas de ECFC, y un mecanismo potencial de aberración microvascular, estrechamiento arteriolar y angiogénesis reducida, descrito previamente en modelos animales48–50. Curiosamente, en niños con pesos al nacer menores de 1.500 g, estos investigadores encontraron una fuerte correlación inversa entre el peso al nacer y los «defectos angiogénicos» de las ECFC (fig. 2A). Este hallazgo es consistente con varios estudios epidemiológicos que muestran una correlación entre el riesgo de HTA en adultos jóvenes y el grado de inmadurez al nacer51,52.
Óxido nítrico y Programación FetalLa retención de sodio en ratas con hipertensión prenatal programada también puede ser el resultado del desequilibrio en la biodisponibilidad del oxido nítrico (NO). En el riñón, el NO cuenta con numerosas funciones importantes como la regulación de la hemodinámica renal, el mantenimiento de la perfusión medular, la modulación de la respuesta tubulo-glomerular y la reabsorción de sodio tubular, lo que resulta en un efecto neto de la natriuresis y la diuresis53.
Cavanal et al.54 midieron la producción de NO en segmentos de aorta de hijos de madres diabéticas. Se observó que la producción basal de NO estaba deprimida significativamente en el grupo de hijos de madres diabéticas en comparación con el control. Tras la estimulación con la acetilcolina (ACh) o bradicinina (BK), la producción de NO aumentó significativamente en todos los grupos, pero el aumento fue de mayor magnitud en los controles. Por otra parte, la disminución renal de angiotensina 1-7 (ANG 1-7) también pueden interferir con la producción y biodisponibilidad del NO, según lo sugerido por Li et al.55. Estos autores observaron que el efecto vasodilatador de la ANG 1-7 en anillos de aorta del ratón fue completamente abolido con el pretratamiento con L-NAME, un inhibidor de la sintasa del NO, indicando que NO endotelial interviene en el efecto vasodilatador de la ANG 1-7 en este modelo experimental.
El mecanismo exacto de la aparición de HTA en los hijos de madres con alteraciones metabólicas durante la gestación no es completamente entendido. Sin embargo, varios mecanismos pueden contribuir al desarrollo de HTA del adulto, reforzando la necesidad de una estrecha vigilancia del metabolismo materno y placentario durante el embarazo para evitar cambios permanentes en la homeostasis de la descendencia.
Bajo peso al nacer y su relación con la función vascular y el estrés oxidativoEs importante destacar que tanto el BPN, como el parto prematuro, inducen cambios en el desarrollo vascular, debido a la inmadurez de varios sistemas biológicos que son modulados por el medio ambiente intrauterino y extrauterino. La importancia de este cambio ambiental, como un evento clave en la etiopatogenia de la disfunción vascular, radica en el crecimiento inapropiado de los vasos sanguíneos que se desarrollan normalmente durante el parto a término. Entre muchas otras diferencias, el medio ambiente intrauterino normal es marcadamente hipóxico en comparación con el medio ambiente extrauterino. A lo largo de la gestación, el feto progresivamente se prepara para la transición hacia el medio ambiente relativamente rico en oxígeno extrauterino, como lo demuestra el aumento considerable de la concentración de enzimas antioxidantes durante las últimas semanas de gestación56. Si el parto ocurre antes de tiempo (particularmente antes de las 32 semanas), esta preparación no se completa, y el feto queda susceptible a factores ambientales como un elevado EO57.
Adicionalmente al estado de EO, la exposición excesiva de glucocorticoides fetales puede aumentar el riesgo de desarrollo de trastornos hipertensivos, confiriendo mayor riesgo de comorbilidad cardiovascular del adulto58. Se ha observado que niños con BPN, en la vida adulta presentan riesgo independiente de sufrir enfermedades psiquiátricas y cardiovasculares, asociadas a desregulación de los niveles placentarios de 11β-HSD (11β-hidroxiesteroide deshidrogenasa)59. Una menor regulación de los niveles de 11β-HSD aumentan la exposición del feto a los glucocorticoides maternos60 y como respuesta adaptativa, se acelera la maduración fetal intrauterina, como fue demostrado recientemente por Roghair et al.61. Adicionalmente, estos autores exploraron los orígenes tempranos de la HTA, en un modelo de rata tratadas con un inhibidor de la 11β-HSD (CBX: carbenoxolona) durante la última semana de gestación. Se demostró que el aumento de la presión arterial se acompañaba de disfunción vascular y producción exagerada de anión superoxido (O2-) en los adultos masculinos, pero no en las mujeres, en las crías expuestas a CBX. Aunque el exceso de glucocorticoides se sabe que inducen la producción de O2- vascular62 y el EO se ha asociado con la disfunción vascular programada63,64, estas interacciones no han sido evaluadas desde una perspectiva de desarrollo.
No obstante, queda aún por responder la pregunta: ¿cómo afecta exactamente este cambio ambiental las características vasculares de niños provenientes de parto prematuro o con BPN? Una posibilidad es que la capacidad angiostática observada en recién nacidos con BPN, sea el resultado de una exposición prematura al medio extrauterino, y dicha capacidad aun no se da. Si es así, se podría especular desde un punto de vista molecular, que el BPN induce defectos en los perfiles de expresión génica que se evidencia en una angiogénesis y un desequilibrio angiostático reducidos. Dichos cambios abarcarían: a) desregulación de moléculas con propiedades angiostáticas como e-NOS y AKT65, b) baja regulación de metaloproteasas de matriz (MMP-2 y MMP-9)66 que degradan la membrana basal y permiten la migración de ECFCs, y c) inapropiada regulación de factores de crecimiento vascular (VEGF, FGF, PGF), incremento de moléculas asociadas con capacidad de proliferación tumoral (PLXDC-1)67 y disminución de citoquinas con actividad angiogénica (CXCL-1 y CXCL-5)68, (fig. 4).
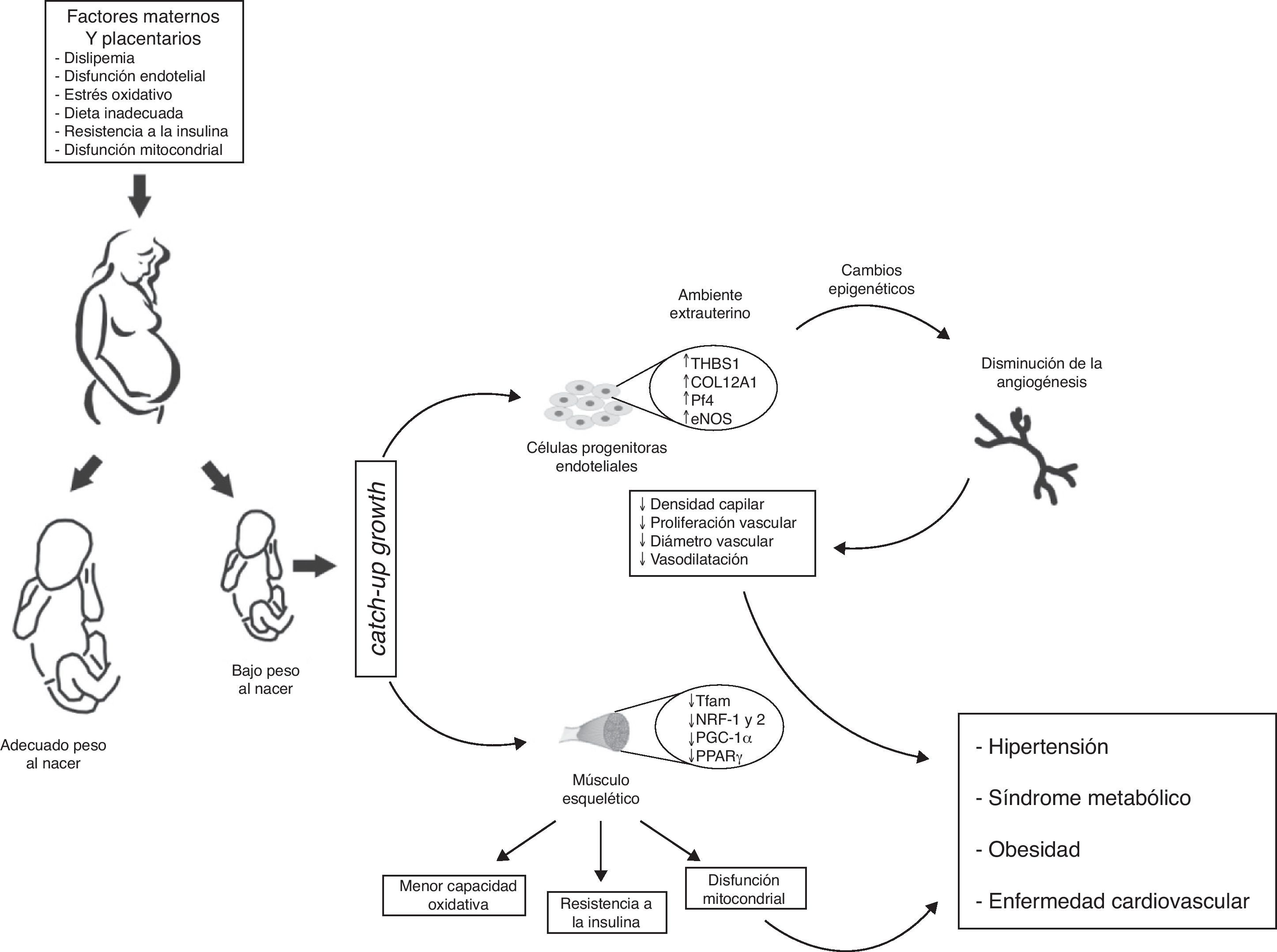
, presentan menor potencial angiogénico (↓ densidad capilar, ↓ diámetro vascular, ↓ proliferación de células endoteliales y ↓ vasodilatación). A nivel vascular esos cambios adaptativos se reflejan en cambios transcripcionales en las rutas metabólicas y del crecimiento vascular. Algunos de estos cambios se obtienen mediante la modificación de la regulación epigenética de los genes (TSBS1, COL12A1, Pf4, eNOS). En músculo esquelético cambios en el ambiente intrauterino (hipoxia fetal) inducen cambios epigenéticos de genes con función metabólica posnatal (Tfam, NRF-1 y 2, PGC-1α, PPARγ), que se traducen en menor capacidad oxidativa, disfunción mitocondrial, y resistencia a la insulina. En consecuencia, infantes con BPN presentan presión arterial más elevada y mayor riesgo de HTA, obesidad, síndrome metabólico y enfermedad cardiovascular en la vida adulta.")
Modelo propuesto de programación de la salud fetal intrauterina y sus consecuencias en la salud de la edad adulta. Las células progenitoras endoteliales provenientes de infantes con bajo peso al nacer (BPN), presentan menor potencial angiogénico (↓ densidad capilar, ↓ diámetro vascular, ↓ proliferación de células endoteliales y ↓ vasodilatación). A nivel vascular esos cambios adaptativos se reflejan en cambios transcripcionales en las rutas metabólicas y del crecimiento vascular. Algunos de estos cambios se obtienen mediante la modificación de la regulación epigenética de los genes (TSBS1, COL12A1, Pf4, eNOS). En músculo esquelético cambios en el ambiente intrauterino (hipoxia fetal) inducen cambios epigenéticos de genes con función metabólica posnatal (Tfam, NRF-1 y 2, PGC-1α, PPARγ), que se traducen en menor capacidad oxidativa, disfunción mitocondrial, y resistencia a la insulina. En consecuencia, infantes con BPN presentan presión arterial más elevada y mayor riesgo de HTA, obesidad, síndrome metabólico y enfermedad cardiovascular en la vida adulta.
En consecuencia, una alteración del patrón de expresión génica es consistente con defectos de la vasculogénesis, falta de anastomosis, menor vasodilatación, mayor proliferación de células musculares lisas y disfunción endotelial. Sin embargo, estos hallazgos no prueban totalmente la relación causal y directa del BPN con los defectos vasculares en la vida posnatal, aunque sí sugieren que contribuyen a la alteración del potencial angiogénico de las células ECFC, como fue demostrado por Ligi I, et al.47 (fig. 4).
Resistencia a la insulina y programación vascular y metabólica del adultoSituaciones que alteran la función vascular durante la vida fetal y neonatal, como hiperglicemia69, diabetes gestacional70,71, resistencia a la insulina72 o hiperoxia73, favorecen el desarrollo de enfermedades cardiovasculares, metabólicas y endocrinas en la vida adulta74–76. Sin embargo, la relación entre un ambiente prenatal inadecuado debido a alteraciones metabólicas maternas, sigue siendo un tema de debate. Amri et al.77 mostraron una disminución del número de nefronas en la descendencia de ratas con diabetes gestacional, tratadas con estreptozotocina (STZ) al inicio del embarazo. En otros estudios, Rocha et al.78 y Magaton et al.79 examinaron el efecto de la diabetes mellitus inducida, en ratas antes del apareamiento, sobre la presión arterial y la función renal de la descendencia. Aunque los resultados mostraron incrementos de las cifras tensionales, no se reportaron cambios en el número de nefronas luego del aislamiento glomerular. Por el contrario, Tran et al.80, mostraron en ratones diabéticos Tg-Hoxb7, que los riñones de crías recién nacidas de madres diabéticas tenían glomérulos con menor área funcional y reducción relativa del número de nefronas. Este resultado está de acuerdo con lo reportado por Ortiz et al.81 al estudiar ratas tratadas con dexametasona en diferentes períodos durante el embarazo, y observar reducción del número glomerular en las crías tratadas.
Además de la aparición de HTA en los hijos provenientes de madres con diabetes gestacional, se ha observado mayor riesgo de obesidad82 y diabetes mellitus tipo 283. Silverman et al.84 evaluaron la descendencia de mujeres con diabetes mellitus pregestacional (tipo 1 y tipo 2) y diabetes gestacional. Se observó que la prevalencia de intolerancia a la glucosa y resistencia a la insulina fue significativamente mayor en estos grupos al ser comparados con el control. Resultados similares fueron obtenidos por Pettitt et al.85, quienes estudiaron el efecto de la tolerancia a la glucosa anormal en la descendencia durante el embarazo de mujeres indígenas PIMA. Los autores correlacionaron las anomalías metabólicas que existen en el embarazo diabético, con estados de resistencia a la insulina, HTA, obesidad y diabetes en los hijos. El mecanismo por el cual la hiperglicemia materna aumenta el riesgo metabólico en los hijos aún no se conoce completamente. Es posible que el aumento en la oferta de de la glucosa para el feto pudiera actuar como un estímulo para mejorar la producción de insulina y exponer al feto a la hiperinsulinemia. Se postula también que el aumento de la producción de leptina fetal puede contribuir al trastorno metabólico en la etapa posnatal86,87. Un aumento de las concentraciones de hormonas como la insulina y la leptina, en períodos críticos del desarrollo, pueden originar un metabolismo «endógeno teratogénico disfuncional»88. Por ejemplo, las crías de ratas hiperglicémicas desarrollan «programación metabólica» de las neuronas del hipotálamo (neuro-peptidérgicas), provocando un aumento de su actividad neuropéptidica y orexigénica, lo cual podría conducir a un estado de hiperfagia y al consecuente aumento de peso89.
Además del efecto de la insulina en el metabolismo de los hidratos de carbono, esta hormona participa en otras funciones, incluyendo la modificación del metabolismo de los lípidos y las proteínas, transporte de aminoácidos e iones, regulación del ciclo celular, apoptosis y síntesis de NO90,91. Además de la insulina, otras hormonas, como la angiotensina II (ANG II) y la norepinefrina, pueden influir en varios procesos biológicos relacionados a la aparición de HTA92,93. La ANG II afecta la señalización de la insulina a través de la vía de las proteínas SOCS-3. Se postula también que la ANG II actúa a través del receptor AT-1 disminuyendo la producción de NO insulino-dependiente, por la inhibición de las proteínas ERK-1/2 y la activación de JNK94. Por otra parte, la ANG II, a través del receptor AT-1, aumenta la actividad de la NADPH oxidasa, incrementando la generación de especies reactivas del oxigeno (ROS)95. Recientemente, Zhou et al.96 demostraron que la regulación de la ANG II y el estrés oxidativo se asocian con disfunción endotelial y resistencia a la insulina en pacientes hipertensos sensibles a la excreción de sodio renal.
La excreción renal de sodio es otro factor importante que contribuye a la aparición de HTA del adulto. Rocco et al.97 estudiaron la excreción de sodio de los hijos diabéticos, con y sin sobrecarga de sodio, y observó que, en condiciones normales, los hijos de madres diabéticas presentaron valores de excreción de sodio similar al grupo control. Sin embargo, luego de la sobrecarga de sodio, los hijos de madres diabéticas no alcanzaron los valores de excreción de sodio del grupo control. De manera semejante, Nehiri et al.98 examinaron la excreción de sodio de hijos de madres diabéticas que recibieron una dieta alta en sodio durante 3 días consecutivos. El estudio de la corteza renal, en los hijos de madres diabéticas, reveló un aumento de la expresión del canal epitelial de sodio (ENAC) y sodio/potasio ATPasa (Na+/K+ ATPasa), sin cambios en el intercambiador de sodio/hidrógeno (NHE3) o de otros transportadores, sugiriendo que la retención de sodio se debía al aumento en la reabsorción de los segmentos distales de la nefrona.
Desarrollo in utero muscular y resistencia a la insulinaLas fibras musculares esqueléticas representan un arreglo molecular heterogéneo, y según su tipo de metabolismo, se denominan fibras tipo I, IIa, IIb y IIx. Estas, a su vez, expresan funciones fisiológicas distintas proporcionadas por la especificidad de sus cadenas de isoformas de miosina99. A comienzos del desarrollo fetal, la mayor expresión de fibras musculares es del tipo I, mientras que las fibras tipo IIa, IIb y IIx se desarrollan en etapas gestacionales más tardías. En modelos animales que presentan el mismo desarrollo embrionario que el humano, (p. ej.: ovejas y cerdos), el principal momento formación de fibras musculares, aparece en la segunda mitad de la gestación, mientras que la expresión de la función metabólica se establece antes del nacimiento100,101. Por lo tanto, sería razonable formular la hipótesis que el músculo esquelético sea un objetivo clave para la programación prenatal, pues un cambio en el ambiente intrauterino podría afectar su composición y la función en la edad adulta. De hecho, varios autores han comprobado que la disminución de las fibras de tipo I y IIa, en sujetos con BPN, se acompaña de una marcada reducción en la capacidad oxidativa muscular, situación que se ha asociado a resistencia a la insulina y obesidad en la etapa adulta102,103 (fig. 4). De forma paralela, Zhu et al.104 observaron que la descendencia de las madres que recibieron una dieta con restricción de nutrientes y calorías, durante el último trimestre de embarazo, presentaron una disminución en el número de fibras musculares y un aumento relativo en el número de fibras tipo IIb en comparación con los hijos alimentados ad libitum.
Recientemente, Thorn et al.105 estudiando un modelo de insuficiencia placentaria crónica, demostraron que los niveles del receptor de insulina en músculo esquelético se incrementaron en un 80%, mientras que expresión de la subunidad catalítica de fosfatidil-inositol 3 quinasa (PI3K) y de la proteína-quinasa-B (Akt/PKB), fueron suprimidos un 36 y 37%, respectivamente. Estos cambios sugieren una estrategia de adaptación metabólica para la supervivencia en casos de RCIU, pero con consecuencias potencialmente perjudiciales en la regulación de los hidratos de carbono y en la sensibilidad a la insulina en la vida posnatal. De hecho, se ha observado que en hijos que han cursado con RCIU, los niveles en musculo de AKT-2, PI3K y GLUT-4, fueron menores en comparación con los hijos con crecimiento normal106. Estos hallazgos sugieren que, a pesar que los fetos con RCIU presentan mayor abundancia en los receptores de insulina en músculo esquelético, el reducido número de moléculas responsable de la activación de los GLUT-4, podría explicar, en parte, la aparición de la resistencia a la insulina y el síndrome metabólico en la edad adulta.
Bioenergética mitocondrial: papel de la mitocondriaLa pérdida del potencial de membrana mitocondrial (Δψm) y la inducción de la permeabilidad transitoria mitocondrial se encuentran estrechamente relacionadas con eventos mitocondriales durante la apoptosis, como evento propio de las células107, y con algunas complicaciones durante la gestación como RCIU, prematuridad y BPN108. La principal consecuencia de la apertura prolongada del poro de permeabilidad transitoria mitocondrial es la despolarización debida a la desaparición del gradiente de protones, y la consecuente inhibición de la respiración mitocondrial. En condiciones de hipoxia fetal, el exceso de ROS genera un aumento en los niveles Ca2+ citosólico, provocando su acumulación en la mitocondria y, con ello, la pérdida del Δψm. Se ha reportado que los ROS pueden disminuir la biogénesis mitocondrial (BM) y alterar el funcionamiento mitocondrial, produciendo disfunción mitocondrial placentaria con el consecuente empeoramiento de la hipoxia tisular y alteraciones del desarrollo placentario109,110. Esto se ha observado a través de la disminución en la expresión de diversos reguladores de la BM, entre ellos, el factor de transcripción mitocondrial A (Tfam) y los factores respiratorios nucleares 1 y 2 (NRF-1 y NRF-2). También en placentas de gestantes con BPN, se han observado cambios mitocondriales bioenergéticos y funcionales111,112. Ali F et al.113 reportaron que, en células endoteliales, la activación del PGC-1α induce la expresión de la hemoxigenasa, una proteína de respuesta al EO. Recientemente, Barrès R et al.114 encontraron una correlación negativa entre la hipermetilación de citosinas (no CpG) del promotor del PGC-1α, y la expresión proteica y la densidad mitocondrial en células musculares de sujetos con diabetes mellitus tipo 2. Adicionalmente, en ratones que recuperan su estado nutricional, hubo una restauración de los niveles de la eNOS y la BM en tejido adiposo y músculo, asociándose además a reducción de peso en comparación con ratones normales115.
Del mismo modo, defectos en la BM y otros relacionados con su morfogénesis, afectan la actividad enzimática y reducen la capacidad oxidativa, conduciendo al aumento de los niveles de lípidos intramiocelulares. Aunado a la incapacidad de la mitocondria para utilizar estos sustratos metabólicos, la acumulación de algunos metabolitos nocivos para su función (p. ej.: ceramidas, diacilglicerol y ROS) en el músculo esquelético, inducen un deterioro en las vías de señalización de la insulina y la consecuente reducción de la captación de glucosa115. Asimismo, alteraciones en las vías de señalización en algunos factores de transcripción que inducen BM, como el PGC-1α y el PPARγ, se han encontrado disminuidos como resultado del cambio en el ambiente intrauterino116,117 (fig. 4). En consecuencia, el deterioro en la captación y oxidación de los ácidos grasos, junto a una menor capacidad antioxidante y un incremento del estado de EO en la vida posnatal, caracterizan la desregulación metabólica en los sujetos con alteraciones en el crecimiento intrauterino. Sin embargo, otros mecanismos, podrían ser modulados por los cambios en el fenotipo celular embrionario, resultado de esta adaptación metabólica.
Perspectivas futurasLas observaciones epidemiológicas de Barker et al.5–7 demostraron tempranamente que adultos que habían tenido un evento coronario o presentaban síndrome metabólico, habían sido pequeños al nacer y permanecieron delgados durante los primeros 3 años de vida. Este retraso del crecimiento fetal, junto al posterior y desproporcionado crecimiento posnatal, se asoció con resistencia a la insulina y disfunción endotelial en la vida adulta. El músculo esquelético es el principal sitio del metabolismo de los hidratos de carbono y de los ácidos grasos, por lo que cualquier aberración en el desarrollo muscular in utero, facilitaría la expresión de enfermedades en la etapa adulta. Asimismo, alteraciones de la distribución de la fibra muscular, cambios en la señalización por la insulina, una reducida capacidad angioneica y una función mitocondrial alterada, son eventos importantes en el desarrollo del enfermedades cardio-metabólicas de los adultos nacidos con BPN, (fig. 4).
El siguiente paso en el camino de las posibles hipótesis de Programación Fetal descritas en esta revisión, seria la comprobación experimental en humanos, en estados metabólicos alterados como preeclampsia, diabetes gestacional, e hipertensión inducida por el embarazo, factores de riesgo asociados a la Programación Fetal de la salud en la edad adulta. También sería interesante explorar algunas vías de señalización relacionadas con la biogénesis mitocondrial (PCG-1α, Tfam, NRF-1 y NRF-2), estado redox (NFKβ) y cambios en la demanda energética (MAPK). En la actualidad, se sabe muy poco de los cambios en la función vascular y la biogénesis mitocondrial en estados metabólicos relacionados con la Programación Fetal de las enfermedades crónicas no transmisibles. Se postula que dicho efecto podría desempeñar un efecto en la atenuación relacionada con la edad mitocondrial y «disfunción» en enfermedades relacionadas al metabolismo oxidativo como miopatías, cáncer y enfermedad mental.
La lactancia materna prolongada, el retaso en la introducción de la alimentación complementaria118, la carga proteica en el primer año de vida, la práctica de ejercicio físico durante la gestación119, etc., son pasos reales sobre los que camina la Pediatría, la Endocrinología y la Gineco-obstetricia de hoy120.
FinanciaciónEl presente estudio no tuvo ninguna fuente externa de financiamiento.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.