


Las mutaciones del gen DAX-1 (DAX1) (gen 1 del cromosoma X, de la hipoplasia adrenal congénita con inversión del sexo, sensible a la dosis; también llamado gen NROB1 [Nuclear receptor subfamily 0, group B, member 1] MIM ID *300473) son responsables del fallo adrenal y del hipogonadismo hipogonadotrópico en pacientes con hipoplasia adrenal congénita (OMIM#300200). Las mutaciones conllevan la pérdida de la expresión de steroidogenic acute regulatory protein (StAR) y LHβ mediada por la represión del steroidogenic factor (1SF-1), así como una reducción de la expresión de GnRH1. Hasta el momento se han descrito más de 100 mutaciones en el DAX11. Es necesario descartar esta entidad clínica en los pacientes que debutan con síndromes pierde sal, cuando las causas frecuentes de fallo adrenal como defectos de la esteroidogénesis (CYP21A2) y alteraciones metabólicas (adrenoleucodistrofia) hayan sido descartadas. Presentamos a 2 hermanos con la mutación (R267P) y heterogenicidad genotípica-fenotípica.
Caso 1Varón que consulta a los 2 meses de vida por cuadro clínico de 15 días de evolución de vómitos y signos de deshidratación, junto con mala curva ponderal. Las determinaciones analíticas revelaron una hiponatremia con hiperpotasemia, junto con un cortisol en rango de normalidad y ausencia de elevación de precursores esteroidogénicos (tabla 1), siendo etiquetado de hipoaldosteronismo primario e iniciándose tratamiento sustitutivo con mineralcorticoides y aporte suplementario de cloruro sódico con adecuada evolución clínica y bioquímica inicial. A los 17 meses de vida fue diagnosticado de déficit asociado glucocorticoideo, ante hiperpigmentación progresiva, estabilidad de la curva de crecimiento con peso percentil (P) 50 y talla P25, e hipocortisolemia. No antecedentes familiares de enfermedades autoinmunes, abortos ni fetos muertos o enfermedades neurológicas, no hermanos en el momento del diagnóstico. Los anticuerpos anti-adrenales y los ácidos grasos de cadena muy larga fueron negativos. La curva de crecimiento fue normal, bajo tratamiento con glucocorticoides 10mg/m2 sc/día y mineralcorticoide 0,175mg/día, hasta que a los 8 años inició una progresiva deceleración con retraso de la maduración ósea. A los 13 años y nueve meses presentaba ausencia de desarrollo de caracteres sexuales secundarios, test combinado hipofisario con ausencia de respuesta de hormona de crecimiento (GH) a la hipoglucemia insulínica (GH basal 0,5ng/ml; pico máximo [pm] 1ng/ml) y al test de clonidina con impregnación esteroidea (GH basal<0,2ng/ml; pm 0,2ng/ml); valores de gonadotropinas y andrógenos sugestivos de hipogonadismo hipogonadotrópico, con test de HCG anómalo (testosterona total basal 0.1ng/ml; pm 1ng/ml). RMN adrenal e hipotálamo-hipofisaria sin hallazgos. A los 14 años, con edad ósea de 10 años, se inició tratamiento con 50mg de propionato de testosterona mensual (4 dosis) no evidenciándose incremento del crecimiento. A los 14 años y 7 meses, con talla 142,6cm (cm/año y edad ósea 10-11 se asoció tratamiento con hormona de crecimiento recombinante (GHr). La velocidad de crecimiento pasó a 8cm/año el primer año con posterior descenso a 6,5-4,7cm/año, alcanzando a los 20 años una talla de 170cm (P25; talla diana 167cm), con caracteres sexuales secundarios adultos. En la actualidad se mantiene tratamiento con GHr ante el déficit de GH del adulto.
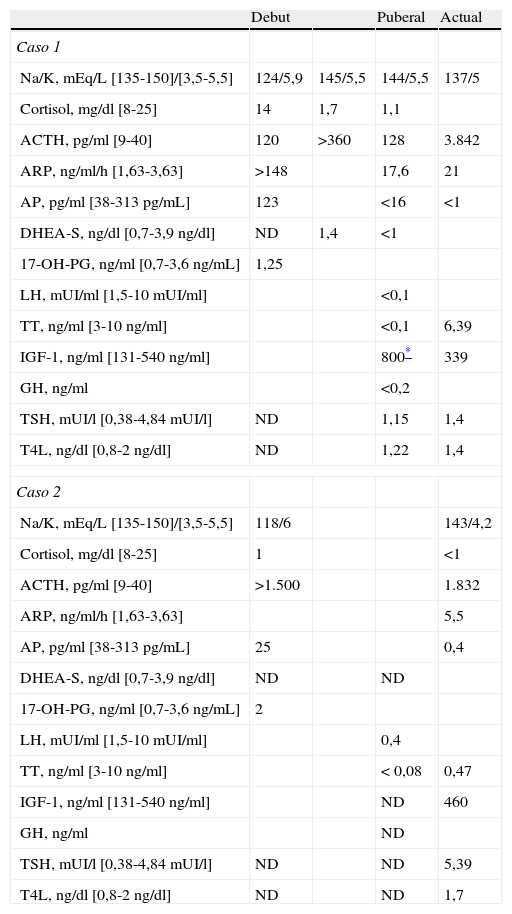
Determinaciones bioquímicas y hormonales basales durante el seguimiento
| Debut | Puberal | Actual | ||
| Caso 1 | ||||
| Na/K, mEq/L [135-150]/[3,5-5,5] | 124/5,9 | 145/5,5 | 144/5,5 | 137/5 |
| Cortisol, mg/dl [8-25] | 14 | 1,7 | 1,1 | |
| ACTH, pg/ml [9-40] | 120 | >360 | 128 | 3.842 |
| ARP, ng/ml/h [1,63-3,63] | >148 | 17,6 | 21 | |
| AP, pg/ml [38-313pg/mL] | 123 | <16 | <1 | |
| DHEA-S, ng/dl [0,7-3,9ng/dl] | ND | 1,4 | <1 | |
| 17-OH-PG, ng/ml [0,7-3,6ng/mL] | 1,25 | |||
| LH, mUI/ml [1,5-10 mUI/ml] | <0,1 | |||
| TT, ng/ml [3-10ng/ml] | <0,1 | 6,39 | ||
| IGF-1, ng/ml [131-540ng/ml] | 800* | 339 | ||
| GH, ng/ml | <0,2 | |||
| TSH, mUI/l [0,38-4,84 mUI/l] | ND | 1,15 | 1,4 | |
| T4L, ng/dl [0,8-2ng/dl] | ND | 1,22 | 1,4 | |
| Caso 2 | ||||
| Na/K, mEq/L [135-150]/[3,5-5,5] | 118/6 | 143/4,2 | ||
| Cortisol, mg/dl [8-25] | 1 | <1 | ||
| ACTH, pg/ml [9-40] | >1.500 | 1.832 | ||
| ARP, ng/ml/h [1,63-3,63] | 5,5 | |||
| AP, pg/ml [38-313pg/mL] | 25 | 0,4 | ||
| DHEA-S, ng/dl [0,7-3,9ng/dl] | ND | ND | ||
| 17-OH-PG, ng/ml [0,7-3,6ng/mL] | 2 | |||
| LH, mUI/ml [1,5-10 mUI/ml] | 0,4 | |||
| TT, ng/ml [3-10ng/ml] | < 0,08 | 0,47 | ||
| IGF-1, ng/ml [131-540ng/ml] | ND | 460 | ||
| GH, ng/ml | ND | |||
| TSH, mUI/l [0,38-4,84 mUI/l] | ND | ND | 5,39 | |
| T4L, ng/dl [0,8-2ng/dl] | ND | ND | 1,7 | |
ACTH: hormona adrenocorticotropa; AP: aldosterona plasmática; ARP: actividad de renina plasmática; DHEA-S: dehidroepiandrosterona sulfato; 17-OH-PG: progesterona; IGF-1: factor de crecimiento insulínico; LH: hormona luteinizante; ND: no disponemos de datos; T4L: T4 libre; TSH: hormona tirotrópica; TT: testosterona total.
Varón diagnosticado a los 18 meses de insuficiencia adrenal por síndrome pierde sal (tabla 1). Dos semanas previas al diagnóstico, tras recibir la vacuna de la DTP y polio oral, el paciente presentó decaimiento y anorexia progresiva, con dolor abdominal nocturno y deposiciones blandas malolientes. Buena ganancia pondero-estatural hasta los 16-17 meses, cuando comenzó con una curva de peso plana, peso<P3, talla P25-50, e hiperpigmentación. Hermano de 8 años con diagnóstico de insuficiencia adrenal primaria (caso1). Los anticuerpos anti-adrenales y los ácidos grasos de cadena muy larga fueron negativos. Desarrollo pondero-estatural normal, bajo tratamiento con glucocorticoides 10mg/m2 sc/día y mineralcorticoide 0,125mg/día, recibiendo suplementos de ClNa vía oral el primer año tras el diagnóstico. A los 12 años se observó una progresiva disminución de la velocidad de crecimiento junto a no progresión del desarrollo puberal. A los 14 años un test combinado hipofisario reveló un eje somatotropo sin alteraciones y un hipogonadismo hipogonadotrópico (LH basal y pm <0,07 mUl/ml, FSH basal y pm 0,6/0,8 UI/l, testosterona total basal <0,08ng/ml). Se instauró propionato de testosterona para inducción puberal (50mg/mes) con incremento del crecimiento, de la maduración ósea y desarrollo progresivo de caracteres sexuales secundarios. A los 17 años el paciente presentaba una talla de 165,5cm (P15), edad ósea 14,5 años, persistiendo velocidad de crecimiento de 6cm/año.
El estudio molecular mediante secuenciación directa del DAX1 con diversas parejas de primers para la amplificación del exón 1, a ambos hermanos y a sus progenitores, mostró que tanto el caso índice (caso 1) como su hermano menor presentaban en hemicigosis un cambio en la posición 800 (c.800G>C), lo que provoca la sustitución a nivel proteico p.Arg267Pro (NR0B1.0004, ARG267PRO). El estudio del ADN de la madre confirmó la presencia de la misma mutación en heterocigosis. El padre no presentó mutación.
La herencia ligada al cromosoma X generalmente es recesiva para las mujeres. En nuestro caso, la madre presenta la mutación en heterocigosis lo que le da la condición de portadora, ya que el alelo dominante normal impide la expresión del gen afecto. Los hijos XY presentan la mutación en hemicigosis y por lo tanto, son enfermos.
Existe una heterogeneidad fenotípica asociada con las mutaciones DAX1. La ausencia de correlación genotípica-fenotípica en ciertas mutaciones es presumiblemente causada por la influencia de otros genes modificadores, conllevando una variación intrafamiliar significativa en la edad de presentación y expresión2. El hermano menor suele debutar a una edad más precoz. La historia familiar suele revelar la presencia de muertes inexplicadas de niños durante su infancia o presencia de hermanos con hipoplasia adrenal congénita. En la mayoría de los casos la forma de presentación es un síndrome pierde sal con hiponatremia, hiperpotasemia y acidosis metabólica en los primeros meses de vida, precedido de la pérdida del canal de crecimiento, pudiendo ser erróneamente diagnosticados de déficit de CYP21A2, hipoaldosteronismo aislado o pseudohipoaldosteronismo2. Mientras que el cortisol basal en el momento del diagnóstico puede variar, la ACTH está invariablemente elevada (caso 1) y existe una inadecuada elevación del cortisol en respuesta al test de estímulo con ACTH. El déficit de aldosterona precede al hipocortisolismo en la mayoría de los pacientes (caso1)3. Están descritos casos de precocidad isosexual transitoria en la infancia y niñez, con elevación de testosterona para su edad, con alargamiento del pene, a veces asociado a un aumento del tamaño testicular, sin otros signos de desarrollo sexual; algunos mecanismos propuestos para este fenómeno implican al gen NR0B1 en el mecanismo de control prepuberal del eje gonadal, el estímulo mediado por la ACTH de la esteroidogénesis testicular o una hiperplasia autónoma de las células de Leydig2. En el período puberal estos pacientes requerirán reemplazamiento con testosterona para el desarrollo de los caracteres sexuales secundarios; dado que las anomalías del DAX1 pueden afectar al desarrollo testicular y la espermatogénesis, el tratamiento de fertilidad mediante GnRH pulsátil y gonadotropinas es a menudo inefectivo4. Pueden a su vez asociar discapacidad mental (motora, lenguaje y comportamiento social). En nuestro conocimiento no existe en la literatura ningún caso reportado que describa la asociación entre DAX-1 y déficit de GH del caso1.
Las mutaciones DAX1 suponen el 58% de los casos de insuficiencia adrenal primaria de «etiología desconocida» en niños (neonatal - 13 años), en los cuales se han descartado causas autoinmunes, defectos en la esteroidogénesis o metabólicas5. A pesar de no cambiar la estrategia terapéutica, el diagnóstico molecular permite el asesoramiento genético en los familiares y estaría justificado en niños que debuten con un síndrome pierde sal de etiología no aclarada, con o sin presencia de déficit de cortisol asociado. Se requiere una elevada sospecha clínica para evitar diagnósticos erróneos y permitir un abordaje terapéutico adecuado.