



Evans’ syndrome is characterized by the reduction of at least two blood cell lines in the absence of other diagnoses; it was previously described as the simultaneous or sequential development of autoimmune hemolytic anemia and immune thrombocytopenia with unknown etiology. Incidence of 37% and mortality rate of 10% have been reported for Evans’ syndrome.
Clinical casesWe report the clinical presentation and evolution of Evans’ syndrome in two infants who were initially diagnosed with immune thrombocytopenia. The clinical diagnosis was supported by complementary studies and hematological disorders were corroborated. Both cases received treatment with steroids and intravenous immunoglobulin.
ConclusionsOther cell line disorders must be looked for when approaching children with thrombocytopenia. In the present cases, we found autoimmune hemolytic anemia and monocytosis. Therefore, infectious and immunological studies must be included. The treatment of choice are steroids, and intravenous immunoglobulin should be considered if severe immune thrombocytopenia is associated, as observed in these cases.
El síndrome de Evans se caracteriza por la disminución de, al menos, dos líneas celulares en ausencia de otros diagnósticos. Anteriormente, se definía como el desarrollo simultáneo o secuencial de trombocitopenia inmune primaria y anemia hemolítica autoinmune sin etiología específica. Se ha reportado una incidencia del 37% y una mortalidad del 10% de este síndrome.
Casos clínicosSe presenta la información clínica y la evolución del síndrome de Evans en dos pacientes lactantes que inicialmente fueron diagnosticados con trombocitopenia inmune primaria. El diagnóstico clínico se apoyó con estudios de gabinete, donde se corroboraron las alteraciones hematológicas. El manejo se realizó con esteroides e inmunoglobulina.
ConclusionesEn el abordaje del paciente pediátrico con trombocitopenia se deben buscar alteraciones de otra línea celular. En los casos reportados se detectó la presencia de anemia hemolítica y monocitosis, por lo que se deben incluir estudios infecciosos e inmunológicos. El tratamiento de primera línea es con esteroides, y debe considerarse la administración de inmunoglobulina si existe trombocitopenia severa asociada, como se observó en estos casos.
Evans syndrome (ES) is defined by the decrease in at least two cell lines in the absence of other diagnoses1. Previously, it was known as the simultaneous or sequential development of primary immune thrombocytopenia and autoimmune hemolytic anemia2. Although few statistical data are available, it has been reported that between 13 and 73% of patients with autoimmune hemolytic anemia (AIHA) have other cell lines affected. In the largest AIHA study, which included 265 children, ES was defined as red blood cell and platelet involvement, and an incidence of 37% and a mortality of 10% were reported.
Specific etiology was identified in only 10% of the patients. ES can be primary or secondary to other diseases, particularly infections, systemic autoimmune diseases or primary immunodeficiencies (which are found in 50 to 60% of cases)1–3. There is only one case reported in the literature associated with influenza vaccination in an adult patient4, and a few cases reported secondary to cytomegalovirus infection5.
This syndrome usually occurs in children between 4 and 12 years old3,6–8. The clinical presentation is characterized by paleness, fatigue, dyspnea, tachycardia, and fever. When thrombocytopenia is present, it is associated with mucocutaneous bleeding. The main hematological alteration of ES is frequently hypochromic, normocytic or macrocytic anemia. Patients may develop gradual or rapid dependence in the range of hemolysis and on the efficacy of the compensatory bone marrow response. Regarding the white line, it is mainly associated with neutropenia1.
Evidence suggesting hemolysis mediated by immunological mechanisms should be sought in the diagnostic approach of ES. These findings are based on the direct positive Coombs test (negative in 5 to 10% of cases), reticulocytes (normal or diminished in up to 40% of cases), indirect hyperbilirubinemia, increased lactate dehydrogenase, and decreased serum haptoglobin; peripheral blood smear may show spherocytes, which suggest extravascular hemolysis. Bone marrow aspiration should be considered when reticulocytopenia is present and in infants with neutropenia. In addition to these tests, an immunological panel with measurements of immunoglobulins and the subclasses of immunoglobulin G (IgG) and, since they can be triggers, serologic and molecular studies should be performed to rule out infectious agents. The first line of treatment is based on the administration of steroids1–3,9. Patients diagnosed with ES should be closely followed because this syndrome has been associated with the development of autoimmune diseases, such as systemic lupus erythematosus, and immunodeficiencies, such as common variable immunodeficiency2,3,7,9.
2Clinical cases2.1Case 1A one-month-three-week-old male infant from Nogales, who did not have hereditary hemato-oncological or perinatal history of importance is described. His family denied exposure to myelotoxics as well as recent vaccination.
Three days before admission, the patient began with the appearance of petechiae on the face with progressive increase and extension of lesions in the thorax, abdomen, genitals, and extremities. Subsequently, wet lesions on the palate and lateral walls of the oral cavity. He arrived to the Pediatric Emergencies Service, where he was found with generalized pallor, conjunctival jaundice, pharynx without secretions, with wet petechiae on the palate and lateral walls of the oral cavity, no palpable adenomegaly, palpable liver at 1 cm below costal border, without splenomegaly, marbled skin, with immediate capillary filling, multiple ecchymosis spots at sites of venipuncture, and generalized and disseminated petechiae. Severe thrombocytopenia was documented, and he was referred to the Pediatric Hematology Department.
The results of the initial blood count (BC) reported normochromic normocytic anemia, monocytosis, and severe thrombocytopenia. A new BC was performed the following day, where a decrease of 1 g / dl hemoglobin, red blood cell distribution width, leukocytosis due to lymphocytosis and monocytosis, severe thrombocytopenia, and normal clotting times were detected. High blood lactate dehydrogenase and phosphorus were found in blood biochemistry. The following results were obtained in an infectious serologic panel: toxoplasma IgG (+) 14.4, IgM (-) 0.05, cytomegalovirus IgG (+) 212, IgM (-) 0.1, human immunodeficiency virus (-). Also, an anemia diagnostic approach protocol was applied, requesting reticulocyte count, which was found to be elevated. Indirect hyperbilirubinemia was found, suggesting hemolysis. Direct Coombs test was negative; however, steroids and immunoglobulin had already been administered. To assess morphology, peripheral blood smears were performed and reported monocytosis, neutropenia, toxic granulations, and reactive lymphocytosis (Table 1).
Case 1 laboratory tests results.
| Initial | Next day | Seven days later | |
|---|---|---|---|
| Hemoglobin | 9.9 g/l with normal indexes | 8.44 g/l with normal indexes | 7.69 g/l with normal indexes |
| EDW | - | 18.4% | 17.4% |
| Leucocytes | 9200 | 16 100 | 8510 |
| Neutrophils | 1800 | 3333 | 3300 |
| Lymphocytes | 5300 | 10 272 | 4000 |
| Monocytes | 1500 | 1964 | 600 |
| Platelets | 11 000 | 11 700 | 349 000 |
| Reticulocytes | - | 6.8% | 9.8% |
| Bilirubin | - | 5.7 | 1.5 |
| DB | - | 0.4 | 0.5 |
| IB | - | 5.3 | 1 |
| LDH | - | 832 | - |
| ALP | - | 333 | - |
| P | - | 6.2 | - |
| Ca | - | 9.9 | - |
| Cl | - | 97 | - |
| K | - | 4.5 | - |
| Na | - | 137 | - |
| Mg | - | 2.3 | - |
EDW, erythrocyte distribution width; DB, direct bilirubin; IB, indirect bilirubin; LDH, lactate dehydrogenase; ALP, alkaline phosphatase.
Imaging studies (abdominal and transfontanelar ultrasound) were reported to be normal.
The patient met the criteria for hemolytic anemia and autoimmune thrombocytopenia (ES). Therefore, immunoglobulin (three doses of 1 g / kg) and methylprednisolone (three doses of 30 mg/kg) were started without blood transfusions.
His clinical condition improved during hospitalization, with excellent response to the treatment and increment in his platelet count (from 11 000 to 231 000 / ml) after the doses of immunoglobulin and methylprednisolone. Given the persistence of findings suggestive of hemolysis, the third dose of immunoglobulin (1 g / kg) was given. After the methylprednisolone doses, hydrocortisone (9 mg/kg/day) was continued and a progressive increase in hemoglobin levels was observed, with normal red blood cell indices, leukocyte decrease with conserved differential cell lines and normal platelet count, as well as improvement in all hemolysis indexes due to reduction of red blood cell distribution width and bilirubin. Only reticulocytosis persisted (Table 1).
Due to conditions of improvement and stability, the patient was discharged with prednisone (2 mg/kg/day). He was followed as an outpatient of the Pediatric Hematology consult. After two weeks, he was found stable and without clinical manifestations suggesting recurrence.
2.2Case 2A 7-month-old male patient, born in Hermosillo, Sonora. His family denied previous hemato-oncological diseases or myelotoxic exposure. Recent vaccination was reported (three weeks before he had received a dose of pentavalent vaccine and influenza). Probable atopic dermatitis was diagnosed three months earlier since he presented generalized rash and an upper respiratory tract infection three weeks before.
One week after receiving the vaccines and having an upper respiratory infection, the patient showed ecchymosis in the occipital region, which extended to his thorax, abdomen, and limbs as well as petechiae and mucosal bleeding (palate, tongue and the lateral walls of the oral cavity). He went to a medical unit where thrombocytopenia was documented (16 000 platelets) and where immunoglobulin, methylprednisolone, and transfusion of platelet concentrates (unknown dosage, frequency, and duration of treatment) were started.
The patient was transferred to our hospital after three days, with already modified clinical manifestations due to the beginning of the treatment and without a previous diagnostic approach protocol.
Upon admission, he presented generalized mucocutaneous bleeding. Severe thrombocytopenia and moderate anemia were corroborated. Pediatric Hematology consultation was requested, and the diagnostic approach protocol started.
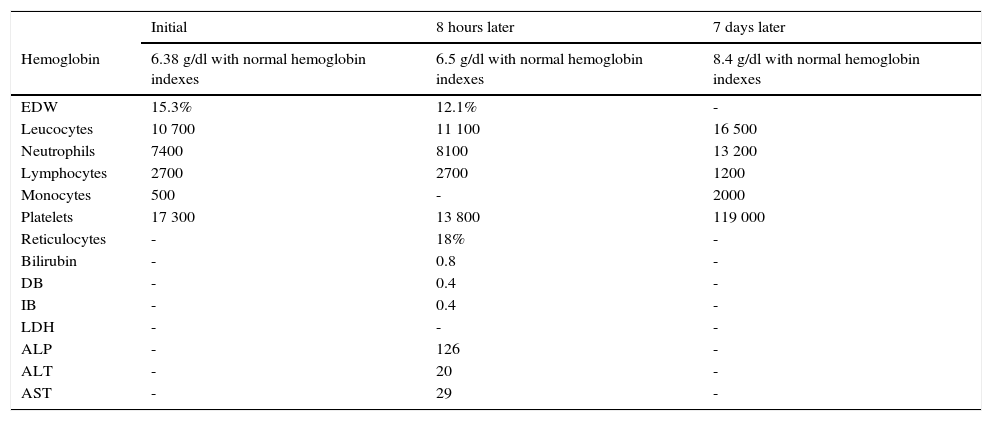
The admission blood analysis reported normochromic normocytic anemia, red blood cell distribution width, conserved white formula, severe thrombocytopenia, and normal serum electrolytes, C-reactive protein and coagulation times. Monitoring was performed during the following 8 hours, without variations of the hemoglobin concentration. Reticulocytosis was corroborated; direct Coombs test was negative, persisted with preserved white formula, severe thrombocytopenia, and normal bilirubin and transaminases (Table 2).
Case 2 laboratory tests results.
| Initial | 8 hours later | 7 days later | |
|---|---|---|---|
| Hemoglobin | 6.38 g/dl with normal hemoglobin indexes | 6.5 g/dl with normal hemoglobin indexes | 8.4 g/dl with normal hemoglobin indexes |
| EDW | 15.3% | 12.1% | - |
| Leucocytes | 10 700 | 11 100 | 16 500 |
| Neutrophils | 7400 | 8100 | 13 200 |
| Lymphocytes | 2700 | 2700 | 1200 |
| Monocytes | 500 | - | 2000 |
| Platelets | 17 300 | 13 800 | 119 000 |
| Reticulocytes | - | 18% | - |
| Bilirubin | - | 0.8 | - |
| DB | - | 0.4 | - |
| IB | - | 0.4 | - |
| LDH | - | - | - |
| ALP | - | 126 | - |
| ALT | - | 20 | - |
| AST | - | 29 | - |
EDW, erythrocyte distribution width; DB, direct bilirubin; IB, indirect bilirubin; LDH, lactate dehydrogenase, ALP, alkaline phosphatase. ALT, alanine aminotransferase, AST, aspartate aminotransferase.
For the study of the triggering etiology, we performed a protocol of infectious agents: TORCH (which reported positive cytomegalovirus IgG of 199) and negative IgM, positive IgG toxoplasma of 7.5 and negative IgM and rubella. A final blood cell cytometry test was performed one day before discharge, with hemoglobin and platelet count improvement, and only with an elevation of the white formula at the expense of neutrophilia and monocytosis secondary to an infectious process in the skin.
In the imaging approach, an abdominal ultrasound was performed, which reported liver and spleen without alterations and of normal size for the patient's age.
The patient presented bicytopenia, defined by normocytic, normochromic regenerative anemia with broad red blood cell distribution width and reticulocytosis. Despite having a direct negative Coombs test and normal bilirubin (probably by modified by the previous initiation of the treatment), he fulfilled the required manifestations to establish the diagnosis of hemolytic anemia and autoimmune thrombocytopenia compatible with ES.
Acute management with immunoglobulin (1 g / kg) and methylprednisolone boluses (mg/kg) was initiated. The patient completed three doses of both, with an adequate response to the management. Clinical improvement was evident from the first 24 hours, although improvement in the laboratory test was slow since hemolysis persisted. Therefore, the third dose of immunoglobulin was administered, and he continued management with intravenous hydrocortisone. During his hospital stay, the patient remained stable and was discharged to continue his outpatient management with prednisone (2 mg/kg) for two more weeks. He was evaluated after two weeks and was found with remission of all previous symptomatology, with normalization of the red formula and platelets and without hemolysis manifestations. However, he presented recurrence of the thrombocytopenia associated with an increase of IgG for cytomegalovirus in two occasions. The patient was evaluated by the Pediatric Infectious Disease Service to continue his follow-up.
3DiscussionFew statistical data on the presentation of Evans syndrome exist. In only 10% of the cases, it is possible to determine an etiology, which is critical for the treatment and prognosis of the disease. In the two reported cases, the association with elevated IgG titers for cytomegalovirus was observed. Although serology is not diagnostic, a suspected latent infection may be confirmed by the polymerase chain reaction (PCR) for cytomegalovirus. This virus can infect immunocompetent patients simultaneously with other agents (such as respiratory syncytial virus, Epstein-Barr virus, Chlamydia pneumoniae, herpes virus 6, measles virus or others10) in up to 68.9% of cases. Therefore, in patients with suspected or diagnosed ES, a complete infectious study should be performed, since cytomegalovirus is the most common infectious agent associated with thrombocytopenia in children between one and six months of age5.
The cases reported in this paper are not in the usual age of presentation of the syndrome, which is between 4 and 12 years3,6–8. In these cases, ES was manifested before one year of age (1 month three weeks and seven months, respectively). In a French cohort, it was showed that 13 of 156 patients diagnosed with ES developed their first-year cytopenia before they were one year old3. The clinical findings in both patients with ES corresponded to the expected according to the affected hematologic cell line and its severity. In these patients, it was manifested as a mucocutaneous hemorrhagic syndrome and anemia syndrome, mainly associated with an infectious syndrome in the first patient. The diagnosis was suspected due to the clinical manifestations and was confirmed by laboratory tests showing the affection of two cell lines (red blood cells and platelets), with evidence of hemolysis. Although direct Coombs test of both patients was negative, the literature mentions this data between 5 and 10% of cases with hemolytic anemia. In these cases, it was not possible to perform the hematological study for the intentional search of erythrocyte antibodies, since our patients had a modified clinical presentation after haemo-transfusion and steroid treatment. It is important to mention that a third hematologic cell line was affected in the first case: monocytes. This fact suggested an infectious etiology of ES as it was associated with elevated IgG antibody titers for cytomegalovirus.
In the reported patients, the IgG value for cytomegalovirus was found to be increased in more than four times its normal value. This led to suspicion of a latent infection; thus, PCR (a more sensitive and specific test) was requested for cytomegalovirus to establish the diagnosis, which resulted negative in both patients. However, the second case has presented two relapses with thrombocytopenia associated with increased IgG against cytomegalovirus5,10,11.
The association of ES with immunodeficiencies should also be considered given that some reports have documented a common variable immunodeficiency associated. Therefore, serum immunoglobulin levels should be determined before starting the treatment with immunoglobulin2,7. Unfortunately, the treatment had already started in both patients; therefore, this study could not be performed at the time of diagnosis.
Furthermore, prior vaccination should be intentionally sought, as its association with the development of primary immune thrombocytopenia (which is a component of the ES), mainly with live attenuated viruses vaccines is well known12,13. In the second patient, a history of influenza vaccination existed; although ES could not be entirely attributed to this factor since an infectious component was also suspected, it should be considered that a case of ES associated with this vaccine has been reported4.
The treatment for both patients was based on immunoglobulin (1 g / kg for three doses) and systemic steroids (methylprednisolone 30 mg/kg for three days), followed by hydrocortisone (8 mg/kg for five days). Prednisone (2 mg/kg) was continued until their next reassessment by the Pediatric Hematology Department. In these cases, it was decided to administer three doses of immunoglobulin (1 g / kg), due to the persistence of laboratory results suggestive of hemolysis, demonstrating an inadequate response to the first line of treatment for autoimmune hemolytic anemia (steroids).
Immunoglobulin is the second line of therapy, with doses ranging from 400 mg to 1 g / kg for 1 to 5 days. Therapeutic regimens for ES are variable and have been adapted from the management of primary immune thrombocytopenia and autoimmune hemolytic anemia.
In the diagnostic approach of the pediatric patient with thrombocytopenia, alterations of another cell line should be sought, since they were initially diagnosed as primary immune thrombocytopenia as observed in both cases. However, there was also red blood cell (hemolytic anemia) and leukocyte (monocytosis) affection, so ES was regarded as a possible diagnosis when clinical and laboratory parameters were met.
The search for an etiology should include infectious and immunological tests, to give an appropriate management in case of determining a specific cause and its adequate follow-up. Treatment will usually be based on steroids as the first line and immunoglobulin if it is associated with severe thrombocytopenia.
It must be remembered that the follow-up of the patient with isolated cytopenia is necessary, since there may be a second cytopenia up to three years later. Therefore, ES should be suspected even in patients who do not refer or who present recurrence.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflict of interestThe authors declare no conflict of interest.
To all the medical (ascribed and scholarship holders) and administrative staff of the Department of Pediatrics of the Hospital General Regional 14 in Ciudad Obregon, Sonora, Mexico.
Please cite this article as: Flores-Montes OA, Escobar-Orduño MC, Lozano-Garcidueñas M, Valle-Leal JG. Síndrome de Evans en lactantes. Bol Med Hosp Infant Mex. 2017;74:141–146.