



Childhood acute leukemia cytological features are unknown in Chiapas, Mexico. Defining these features is important because this is a relatively isolated population with high consanguinity index, and these aspects could determine differences in the response to treatment and outcome.
MethodsEighty-one childhood acute leukemia cases treated at the Hospital de Especialidades Pediátricas (HEP) in Chiapas were characterized by morphology, immunophenotype, genotype, initial risk assignment and status at the time of the study. Results were compared with national and international relevant information.
ResultsThe following proportion of acute leukemia types was found: B-cell, 75.3%; myeloid, 16%; T-cell, 3.7%; B-M, 3.7% and NK cells, 1.2%. Genetic alterations were present in 40.6% of B-cell and 69% of myeloid. The genetic alteration was related to the patient's short-term evolution in type B leukemias but not in myeloid. In B-cell leukemia, the cases with the altered MLL gene died in less than one month; cases with t(1;19) (q23;p13) translocation had a good evolution, and those with t(12;21) (p13;q22) translocation had a poor evolution in the medium term. Hyperdiploidy occurred in 20% of B-cell cases; 83% of them remained in remission 1 to 12 months after diagnosis and 69% of the cases with myeloid leukemias died or abandoned the treatment at relapse after 15 days to 37 months of diagnosis.
ConclusionsThe proportion of different types of acute leukemia treated at the HEP is similar to that found in other parts of the country. Their behavior and outcome are related to the presence or absence of specific and non-specific genetic alterations.
Se desconocen las características citopatológicas de las leucemias agudas en pacientes de Chiapas, México, ya que es una población relativamente aislada con alto índice de consanguinidad, lo cual podría afectar la evolución y la respuesta terapéutica.
MétodosSe clasificaron morfológica, inmunofenotípica y genotípicamente 81 casos de leucemia aguda en pacientes atendidos en el Hospital de Especialidades Pediátricas de Chiapas (HEP), indicando riesgo al ingreso y situación al momento del estudio. Los resultados se compararon con información nacional e internacional pertinente.
ResultadosSe encontró la siguiente proporción de tipos de leucemia aguda: leucemias B, 75.3%; mieloides, 16%; de células T, 3.7%; B-M, 3.7% y de células NK, 1.2%. Las alteraciones genéticas estuvieron presentes en 40.6% de las B y en 69% de las mieloides. En las B, la alteración genética se relacionó con la evolución del paciente a corto plazo, no así en las mieloides; además, en las B, los casos con el gen MLL alterado fallecieron en menos de un mes. Los pacientes con la translocación t(1;19)(q23;p13) han tenido buena evolución, no así con la t(12;21)(p13;q22), en la cual la evolución ha sido mala a mediano plazo. La hiperdiploidía se presentó en 20% de los casos B; 83% de ellos permanecen en remisión de 1 a 12 meses desde el diagnóstico. El 69% de los casos con leucemias mieloides falleció o abandonado tratamiento en recaída de 15 días a 37 meses desde el diagnóstico.
ConclusionesLa proporción de los diferentes tipos de leucemia aguda atendidas en el HEP es similar a la encontrada en otras partes del país. Su comportamiento y desenlace está relacionado con la presencia o ausencia de alteraciones genéticas específicas y no específicas.
Since the creation of the Popular Health Insurance in 2004,1 most commonly known as Seguro Popular, indigenous people from Chiapas have been subscribing to this program that provides free diagnosis and complete treatment for some diseases, neoplastic diseases in children among them. In order to satisfy the growing demand for medical care derived from the Seguro Popular, in 2006 the Centro Regional de Alta Especialidad was created, constituted by the Hospital de Especialidades Pediátricas (HEP) de Tuxtla Gutiérrez, to attend to the pediatric population, and the Hospital Regional de Alta Especialidad en Ciudad Salud, Tapachula, for the adult population.2
With this scheme of health care services, it is now possible to know more accurately the health problems that affect historically isolated indigenous groups.
Among the emerging diseases in the pediatric population, neoplastic diseases are notable for their impact. Since the beginning of HEP activities in 2006, these have been the group of diseases for which patients are most frequently hospitalized, and among them, acute leukemia (AL) has been the more frequent diagnosis at hospital discharge according to the biostatistics area of the HEP.
The precise diagnosis of AL is currently based on two main pillars: clinical and cytopathological characterization of malignant blood cells both in peripheral blood and bone marrow. This includes the quantification and microscopic observation of its characteristics, immunophenotype and genetic studies (DNA index, karyotype, and molecular translocations) in bone marrow samples. To date, there is no detailed report of these characteristics in the population of patients with AL treated in the HEP.
Since late 2013 (date on which a flow cytometer was available at the HEP), it became possible to carry out the study of suspicious cases of leukemia in the hospital, and to define more accurately the immunophenotypic characteristics of the malignant cells (i.e., the cell line and subpopulation affected, their degree of differentiation and the expression of aberrant markers). Subsequently, advances in the knowledge and study of the human genome in various pathologies have identified different, specific and non-specific karyotypic translocations and alterations and their relation to different types of leukemia (defined by immunophenotype). The sum of these elements has led to the identification of subsets of leukemias within the same cell line, allowing the cytopathological, prognostic and therapeutic individualization. As a result, it is possible to assign a specific treatment—defined by the National Application Protocols—to each patient.3,4
In this cross-sectional retrospective study, the results of the detailed cytopathological (morphological, immunophenotypic and genotypic) characterization of 81 cases of AL in children, treated in the HEP from 2013 to 2016 in a mostly indigenous population of the state of Chiapas are presented. The above with the objective of having initial panoramic information of the subtypes of AL affecting the pediatric population treated at the HEP (study group). Additionally, information about the various types of AL identified as well as the clinical status of the patients at the time of this analysis and their initial risk classification was presented. The findings were compared with those obtained in other national and international centers.
2MethodsEighty-one consecutive cases of AL were evaluated between December 2013 and June 2016. The immunophenotype was performed at the HEP cytometry laboratory. These accounted for approximately 20% of the total cases seen in the HEP since the beginning of its activities. Additional data was obtained from the electronic database of the HEP laboratory (Athenea), the patient's current evolution and situation, and the electronic clinical records of the Hospital Management Information System (SIGHO) of the HEP.
2.1DiagnosisIn all cases, the diagnosis of AL was performed by a pediatric hematologist/oncologist of the HEP and supported with the compatible clinical picture (blasts in circulating blood, more than 30% of blasts in bone marrow aspirate). Preliminarily, the affected cell line was classified in lymphoblastic (L1, L2 or L3) or myeloblastic (AML), according to the morphological appearance of blasts.
The leukocyte count in peripheral blood was performed using an electronic cell counter (Advia 120, Siemens Healthcare Diagnostics, Eschborn, Germany) following the manufacturer’s recommendations. Blood and bone marrow smears were stained with Wright (generic) staining and analyzed conventionally, making an initial plausible diagnosis and a provisional classification of leukemia according to the morphological characteristics of the blasts.
2.2ImmunophenotypeIn 67 of the 81 cases (83%), the immunophenotyping was performed as a diagnostic study, and in 14 cases (17%), it was performed in previously known patients at the time of relapse (secondary). In all cases, immunophenotyping was performed on the bone marrow sample using BD FACSCanto a FACScan II (Becton Dickinson and Co., New Jersey, USA) equipped with red (633nm) and blue (488nm) lasers and conjugated antibodies to fluorochromes [fluorescein isothiocyanate (FITC), phycoerythrin (PE), peridin chlorophyll protein (PerCP-Cy5.5) or allophycocyanin (APC)]. Direct cell staining was used following the procedure recommended by the manufacturer. Marker panels were designed internally, trying to include recommended antibodies by the consortium EuroFlow.5 The study of each case was carried out in two rounds: in the first, the existence of a clonal neoplastic population and the cell line involved were determined using three tubes, each with a combination of four antibodies; in the second round, the neoplastic subpopulation and its degree of differentiation were determined. The coexistence of other neoplastic lines was ruled out, and the expression of aberrant markers was evaluated. A minimum of seven tubes was used per case, each with four antibodies (28 determinations). The conjugated antibodies employed were directed against the following antigens: CD1a, CD2, CD3, CD4, CD5, CD7, CD8, CD10, CD11b, CD13, CD14, CD15, CD16, CD20, CD22, CD34, CD36, CD38, CD45, CD45, CD56, CD58, CD61, CD64, CD71, cCD79a, CD117, CD123, CMPO (cytoplasmic myeloperoxidase), nTdT (deoxynucleotidyl transferase nuclear terminal), HLA-DR (human leukocyte antigen-DR), CIGM k, l CIGM, TCR α / β TCR and γ / δ. Typically, the assessment of one case of B leukemia included 19 different specificities in 28 determinations; T leukemia assessment included 21 specificities, and myeloid leukemia assessment included 20 specificities. In the case of the latter, the study usually extended, reaching up to 36 specificities in 40 determinations. A marker was considered positive when it reacted with 20% or more of the studied cells. Results were analyzed with the BD program FACS Diva v.6.1.3 (BD Biosciences, Becton Dickinson, and Co., New Jersey, USA).
This analysis confirmed the initial diagnosis (clinical and laboratory) and the neoplastic cell line was accurately determined. Cases were sub-classified based on the modified guidelines of the European Group for Immunologic Classification of Leukemia (EGIL) according to their aberrant expression of antigens, depending on their stage of differentiation or the affected subpopulation.6,7 AML cases initially assigned to the various subgroups of the French-American-British (FAB) classification by morphology were ratified or corrected according to the detailed immunophenotype, following the guidelines of Abdul-Hamid.8 For the sub-classification of mixed phenotypes AL (MPAL; bilinear or biphenotypic) the EGILS’ Criteria Rating System for Acute Leukemia Biphenotype was followed.6
2.3Translocations and other gene rearrangementsBone marrow samples were analyzed for the presence of the 26 most common translocations in AL: inversion and deletion of DOT1L (DOT1-like, disruptor of telomeric 1-like). The RT-PCR commercial nested multiplex (Hemavision ® -28N; DNA Diagnostic, Risskov, Denmark) was used following the manufacturer's instructions.
2.4KaryotypeIn 73 cases, karyotype analysis of bone marrow cells was performed, obtaining suitable metaphases in 68 of the 73 studied cases. The karyotype analysis was carried out using a conventional technique of non-stimulated cell culture for 24 to 48h, followed by addition of colchicine, hypotonic solution and fixative solution, and further G-band staining and microscopic analysis.
2.5DNA indexIn all cases, the DNA index was determined in bone marrow cells with flow cytometry using the Coulter ® DNA Prep™ kit for extraction and DNA staining and Epics XL ® cytometer (both of Beckman Coulter, Brea, CA, USA).
After obtaining the genetic studies results, EGIL subtypes were correlated with the third version of the International Classification of Diseases-Oncology, of the World Health Organization (WHO) (ICD-O-3).9
2.6EthnicityThe determination of ethnicity was carried out subjectively, considering the degree of marginalization and size of the locality of origin, the dominant language of the parents and the self-assessment of the relatives responsible for the patient.
3Results3.1General characteristicsThe mean age of the patients was seven years eight months (95% CI 6.7-8.7 years). The distribution of cases by age group showed 31 cases (38.3%) in children younger than 5 years old, 24 cases (29.6%) in children between 5 and 9 years old, 20 cases (24.7%) in children between 10 to 14 years old, and 6 cases (7.4%) in adolescents between 15 to 18 years old. The male/female ratio of the cases was 1.3: 1, similar to the 1.4:1 ratio observed in non-leukemic hospitalized patients at the HEP during the same period.
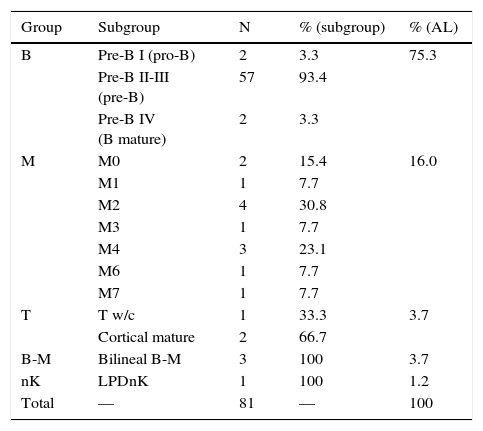
3.2Classification and sub-classificationTable 1 presents the definitive diagnoses of the patients in order of frequency and subgroup according to EGIL. Leukemias affecting B-lymphocytes predominated (75%), followed by myeloid leukemias (16%), mixed phenotype (4%), T lymphocytes (4%) and isolated cases from other varieties. In all secondary cases, the study matched the primary diagnosis (performed by an external provider).
Classification of patients with acute leukemias.
| Group | Subgroup | N | % (subgroup) | % (AL) |
|---|---|---|---|---|
| B | Pre-B I (pro-B) | 2 | 3.3 | 75.3 |
| Pre-B II-III (pre-B) | 57 | 93.4 | ||
| Pre-B IV (B mature) | 2 | 3.3 | ||
| M | M0 | 2 | 15.4 | 16.0 |
| M1 | 1 | 7.7 | ||
| M2 | 4 | 30.8 | ||
| M3 | 1 | 7.7 | ||
| M4 | 3 | 23.1 | ||
| M6 | 1 | 7.7 | ||
| M7 | 1 | 7.7 | ||
| T | T w/c | 1 | 33.3 | 3.7 |
| Cortical mature | 2 | 66.7 | ||
| B-M | Bilineal B-M | 3 | 100 | 3.7 |
| nK | LPDnK | 1 | 100 | 1.2 |
| Total | — | 81 | — | 100 |
AL, acute leukemia; w/c, without classification; LPD, lymphoproliferative disorder.
According to their degree of differentiation, B-cell acute lymphoblastic leukemia (B-ALL) was sub-classified in pre-B I to IV,7 the corresponding pre-B I to pro-B, pre-B II and III to the pre -B common or pre-B, and pre-B IV to mature B (total 61/81, 75%).
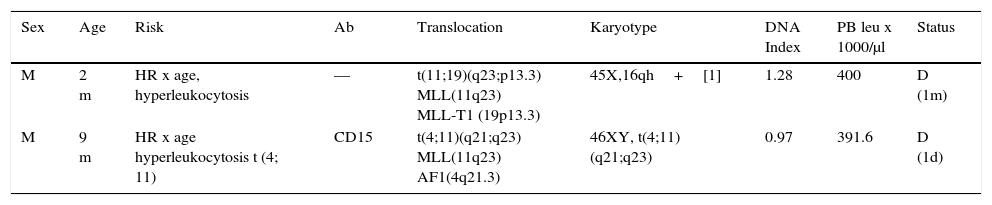
Pre - B I (pro - B; 2/61). Two ALL pro - B cases were identified and characterized by immunophenotype (CD34+, CD45 int, nTdT+, CD19+, CD22+, HLA-DR+, CD38+, cCD79a+, CD10-, CD20-, cIgM-) (Table 2). In this series, the pro-B ALL represented 3.3% of cases of B-ALL and 2.9% of all lymphoblastic leukemias. Both cases occurred in male patients younger than one year old. In both cases, an 11q23 rearrangement in the MLL gene was detected; one of them with a t (11; 19) (q23; p13.3) (MLL - T1) translocation and the other with a t (4; 11) (q21; q23) (MLL - AF1) translocation, corresponding to the WHO subgroup of leukemia / lymphoma with MLL gene rearrangement by translocation t (v; 11q23); MLL (ICD-O-3; 9813/3). These cases had marked leukocytosis with an initial count>300,000 / μl. One showed an aberrant expression of CD15, and the other was hyperploid (DNA index>1.16). Both patients died in the short-term after diagnosis.
Pre-BI (pro-B) leukemia.
| Sex | Age | Risk | Ab | Translocation | Karyotype | DNA Index | PB leu x 1000/μl | Status |
|---|---|---|---|---|---|---|---|---|
| M | 2 m | HR x age, hyperleukocytosis | — | t(11;19)(q23;p13.3) MLL(11q23) MLL-T1 (19p13.3) | 45X,16qh+[1] | 1.28 | 400 | D (1m) |
| M | 9 m | HR x age hyperleukocytosis t (4; 11) | CD15 | t(4;11)(q21;q23) MLL(11q23) AF1(4q21.3) | 46XY, t(4;11)(q21;q23) | 0.97 | 391.6 | D (1d) |
Two cases of B-ALL 61 (3.3%), (WHO ICD-O-3; 9813/3), CD34+immunophenotype, CD45-, nTdT+, CD19+, CD22+, cCD79a+, CD10-, CD20-, cIgM-.
WHO ICD-O-3, World Health Organization, International Classification of Diseases-Oncology-Version 3; Ab, aberrant; leu, leukocytes; PB, peripheral blood; M, male; HR, high risk; RR, regular risk; D, death.
Pre-B II-III (common pre-B or pre-B; 57/61). Of the 61 B-ALL cases, 57 (93%) of them had an immunophenotypic leukemia pre B II – III (or common ALL pre-B) profile (CD34+/ -, CD45- / int, nTdT+/ -, CD19+, CD10+, CD20+/-, CD22+, HLA-DR+, cCD79a+/-, cIgM+/-, κ or λ -) from the earliest (pre-B II; CD34+, cIgM -) to the latest in the differentiation (pre - B III; CD34-, cIgM+), but without the expression of IgM on the surface. These cases were divided into those with genetic alterations (22/57; 39%) and those without them (35/57; 61%).
Pre-B with genetic alterations (22/57). In cases with genetic alterations, three variants were identified: cases with structural changes (translocations and others), with numeric abnormalities (hyperdiploidy and hypodiploidy) and with both.
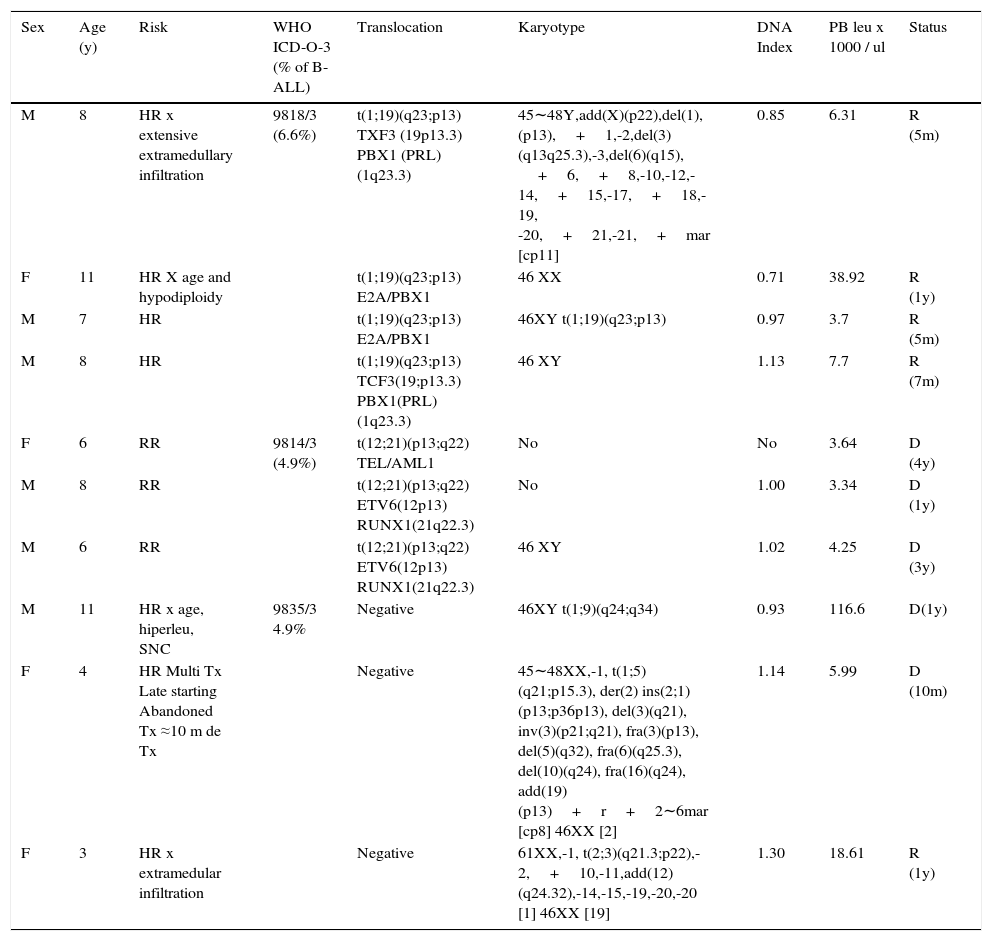
Translocations identified by RT-PCR or karyotype (10/22). There were six cases detected by RT-PCR, three by karyotype and one by both methods for a total of 10 cases (17.5% of pre B cases; 16.4% of B-ALL; 14.7% of ALL) (Table 3). The most common translocation was t (1; 19) (q23; p13) (sub group ICD-O- 3; 9818/3), which was present in four CD34 negative cases (6.6% of B-ALL). All of them had a favorable evolution and were in remission after 5 to 12 months since diagnosis, two of them despite having a high risk (one due to age and one due to hypodiploidy and extensive initial extramedullary disease). Three cases (5% of B - ALL) presented t (12; 21) (p13; q22) (WHO ICD-O-3 subgroup; 9814/3). All died 1 to 4 years after the relapse diagnosis, despite having a regular risk at the beginning and a favorable response to the initial treatment. Three cases had translocations not considered in the WHO classification: one presented the t (1; 9) (q24; q34) translocation and an aggressive evolution with a second relapse in the central nervous system (CNS), and passed away within a year. The other presented the t (1; 5) (q21; p15.3), and abandoned treatment 10 months after diagnosis and died. The last case in this group presented t (2; 3) (q21.3; p22) and hyperdiploidy and has evolved favorably. One year after diagnosis, this patient remains in remission despite an initial extensive extramedullary infiltration.
Pre-B leukemia, CD10 positive (common) with translocations.
| Sex | Age (y) | Risk | WHO ICD-O-3 (% of B-ALL) | Translocation | Karyotype | DNA Index | PB leu x 1000 / ul | Status |
|---|---|---|---|---|---|---|---|---|
| M | 8 | HR x extensive extramedullary infiltration | 9818/3 (6.6%) | t(1;19)(q23;p13) TXF3 (19p13.3) PBX1 (PRL)(1q23.3) | 45∼48Y,add(X)(p22),del(1),(p13),+1,-2,del(3)(q13q25.3),-3,del(6)(q15), +6,+8,-10,-12,-14,+15,-17,+18,-19, -20,+21,-21,+mar [cp11] | 0.85 | 6.31 | R (5m) |
| F | 11 | HR X age and hypodiploidy | t(1;19)(q23;p13) E2A/PBX1 | 46 XX | 0.71 | 38.92 | R (1y) | |
| M | 7 | HR | t(1;19)(q23;p13) E2A/PBX1 | 46XY t(1;19)(q23;p13) | 0.97 | 3.7 | R (5m) | |
| M | 8 | HR | t(1;19)(q23;p13) TCF3(19;p13.3) PBX1(PRL) (1q23.3) | 46 XY | 1.13 | 7.7 | R (7m) | |
| F | 6 | RR | 9814/3 (4.9%) | t(12;21)(p13;q22) TEL/AML1 | No | No | 3.64 | D (4y) |
| M | 8 | RR | t(12;21)(p13;q22) ETV6(12p13) RUNX1(21q22.3) | No | 1.00 | 3.34 | D (1y) | |
| M | 6 | RR | t(12;21)(p13;q22) ETV6(12p13) RUNX1(21q22.3) | 46 XY | 1.02 | 4.25 | D (3y) | |
| M | 11 | HR x age, hiperleu, SNC | 9835/3 4.9% | Negative | 46XY t(1;9)(q24;q34) | 0.93 | 116.6 | D(1y) |
| F | 4 | HR Multi Tx Late starting Abandoned Tx ≈10 m de Tx | Negative | 45∼48XX,-1, t(1;5)(q21;p15.3), der(2) ins(2;1)(p13;p36p13), del(3)(q21), inv(3)(p21;q21), fra(3)(p13), del(5)(q32), fra(6)(q25.3), del(10)(q24), fra(16)(q24), add(19)(p13)+r+2∼6mar [cp8] 46XX [2] | 1.14 | 5.99 | D (10m) | |
| F | 3 | HR x extramedular infiltration | Negative | 61XX,-1, t(2;3)(q21.3;p22),-2,+10,-11,add(12)(q24.32),-14,-15,-19,-20,-20 [1] 46XX [19] | 1.30 | 18.61 | R (1y) |
Ten cases of B-ALL 61 (16.4%), CD34+/- immunophenotype, CD45-, nTdT+, CD19+, CD10+, CD20+/-, CD22+, cCD79a+/-,+/- CIGM.
WHO ICD-O-3, World Health Organization, International Classification of Diseases-Oncology-Version 3; leu, leukocytes; PB, peripheral blood; M, male; F, female; HR, high risk; CNS, central nervous system; RR, regular risk; Tx, treatment; R, remission; D, death; Hiperleu, hyperleukocytosis; m, months; y, years.
Only karyotype alterations (12/22). Table 4 presents 12 cases (21% of pre–B; 20% of B – ALL cases; 15% of ALL cases) that presented only karyotype alterations (cases that also presented translocations were excluded). In this group, six cases were initially classified as HR, by age or disease with extensive extramedullary infiltration. Two cases presented hyperleukocytosis >50 000 / μl. Eight of the cases had hyperdiploidy: seven of them with a DNA index >1.16 and one by karyotype >50 chromosomes (ICD-O-3 subgroup; 9851/3). Four additional cases had various karyotype alterations (ICD-O-3; 9835/3). Although initially classified as HR (six patients) and having many karyotype alterations, 10 out of 12 cases in this group were in remission (4 months to a year after diagnosis). One patient died a month after diagnosis and another was transferred to a different institution. None of them had hypodiploidy.
Pre-B leukemia, CD10 positive (common) with karyotypic alterations.
| ex | Age (y) | Risk | WHO-ICD-O-3 (% of B-ALL) | Karyotype | DNA Index | PB leu x 1000 / ul | Status |
|---|---|---|---|---|---|---|---|
| M | 3 | RR | 9815/3 (13.1%) | Hyperdiploid (> 50) [2] 46XY [18] | 0.93 | 3.28 | R (5m) |
| M | 4 | HR x renal infiltration | 46XY [18], 48XY,+16,+18 [1], 56XXY,+5,+6,+7,+8,+10,+17,+18,+21,+21 [1] | 1.20 | 1.88 | R (1y) | |
| M | 2 | RR | 46XY | 1.36 | 31.84 | R (9m) | |
| M | 2 | RR | 46XY | 1.25 | 0.95 | R (9m) | |
| F | 7 | HR x mediastinal infiltration | 46XX | 1.19 | 3.56 | R (1y) | |
| M | 3 | RR | 46XY | 1.19 | 9.19 | R (4m) | |
| M | 1 | HR x age and leu>50,000 | 46XY | 1.4 | 86.18 | R (6m) | |
| F | 5 | HR x leu>50,000 | 46XX | 1.17 | 66.53 | R (7m) | |
| M | 4 | RR | 9835/3 (6.5%) | 44XY,+1,9qh+, - 14, -17, -20 [1] 45XY, 9qh+,+10, -13, -19 [1], 45X, 9qh+[1], 46XY 9qh+[9] | 1.14 | 23.91 | R (1y) |
| F | 16 | HR x age | 46XX -20,+sea [2] 46XX [18] | 1.02 | 3.9 | D (1m) | |
| M | 15 | HR x age | 46XY, -20,+sea [2] 46XY [18] | 1.05 | 5.22 | R (10m) | |
| M | 4 | RR | 46XY, the (2) (q12; q21.3) [1] | 1.00 | 7.76 | T (IMSS) |
Twelve of 61 cases of B-ALL (6.19%) without translocations detected with CD34+immunophenotype, CD45-, nTdT+, CD19+, CD10+, CD20+/-, CD22+, HLA-DR+, cCD79a+/-,+/- CIGM.
WHO ICD-O-3, World Health Organization, International Classification of Diseases-Oncology-Version 3; ALL, acute lymphoblastic leukemia; leu, leukocytes; PB, peripheral blood; M, male; F, female; RR, regular risk; HR, high risk; R, remission; D, death; T, transferred; m, months; y, years; IMSS: Mexican Institute of Social Security.
Pre - B without genetic alterations (35/57). The group of pre-B ALL without genetic alterations was the largest (subgroup 9811/3 ICD-O-3). Of all 35 cases (61% of pre-B ALL, 57% of B-ALL), 21 were male and 14 female, with a mean age of 7.9 years; 18 were initially classified as HR or VHR (very high risk). Aberrant expression of CD13 was found in four patients; three patients had aberrant expression of CD36 and one of CD33. Seven of the 35 patients died within one month and four years after diagnosis. Twenty-four remained in remission 2 to 19 months after diagnosis and four had abandoned treatment.
B-ALL (mature; 2/61). In the B-ALL group, two cases of mature B-ALL were identified (CD34 -, CD45- / int, nTdT -, CD19+, CD10+/ -, CD20+, CD22+, HLA-DR+, cCD79a+/-, cIGM+, κ+), characterized by the presence of IgM on the cell surface with light in the clonal neoplastic population with k chains (ICD-O-3 subgroup; 9826/3). Both cases were classified with regular risk; one of them presented hyperdiploidy (DNA index 1.34) and 46XY, 21PS+ karyotype. No translocations were found, and both were in remission three and nine months after diagnosis, respectively. c-Myc alterations were not determined. Both cases were treated with the ALL protocol.
3.2.2Acute myeloblastic leukemiaIn the present series, 16% of the cases (13/81) of AL treated at the HEP corresponded to AML. In these cases, patients gender ratio was 1.6: 1 (female/male), and mean age was slightly higher than common for ALL cases (9.3 vs. 7.9 years old), with no significant difference (p>0.05 determined by unpaired t-test).
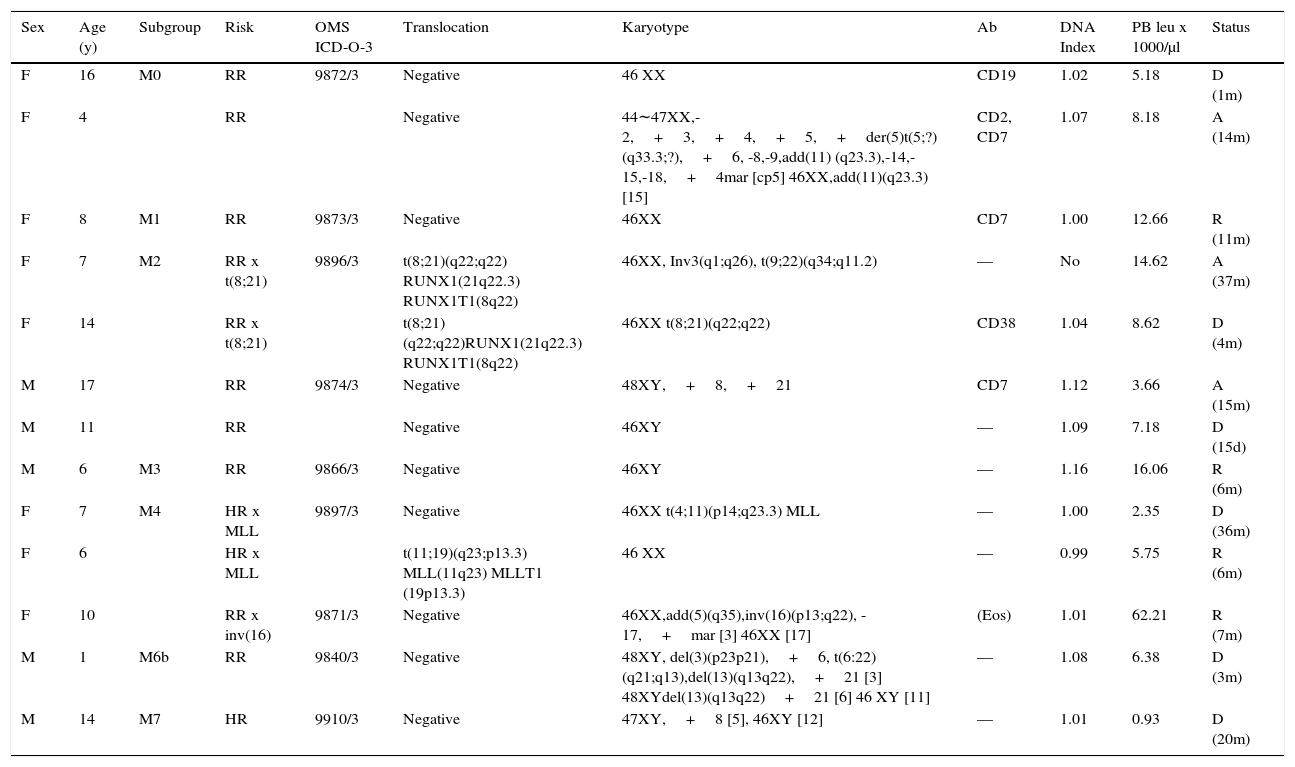
Table 5 presents the sub-classification of myeloid cases describing their initial risk assessment and their status at the time of the current report. Nine of the 13 patients (69%) had genetic translocations, karyotype alterations or both. Of these patients, 6/9 died or abandoned the treatment and in relapse (3/9) 15 days to 37 months from the diagnosis, without a clear relation to the genetic profile. Seven had an initial regular risk. Only four of the 13 cases were in remission 6 to 11 months after diagnosis, one of them was initially classified with HR. Both patients (M2) that presented the (t8; 21) (q22; q22) translocation evolved unfavorably. One died after four months and the second progressed to chronic myeloid leukemia with positive Philadelphia chromosome—with the translocation t (9; 22) (q34; q11.2) BCR-ABL1—for which the patient received imatinib and nilotinib plus hydroxyurea with favorable temporal responses. The patient abandoned treatment in M2 relapse after 37 months. One M4 case with MLL gene alteration (11q23.3) due to t (4; 11) (p14; q23.3) died 36 months after diagnosis. The other M4 case with alteration of the MLL gene due to t (11; 19) translocation (q23; p13.3) continued in remission six months after diagnosis. The third patient with M4 presented with eosinophilia and inversion inv (p13; q22); it was the only case in the series with hyperleukocytosis of >50,000 / μl and in remission seven months after diagnosis. The M6 case with t (6:22) (q21; q13) translocation and other karyotype alterations died three months after diagnosis. Finally, an M7 case persisted with severe bicytopenia (anemia and leukopenia) and moderate thrombocytosis for several months without receiving antineoplastic treatment, who presented a myeloblastic crisis and died. Bone marrow and peripheral blood showed blasts with an M7 phenotype (CD34+, CD36+, CD61+).
Myeloid leukemia.
| Sex | Age (y) | Subgroup | Risk | OMS ICD-O-3 | Translocation | Karyotype | Ab | DNA Index | PB leu x 1000/μl | Status |
|---|---|---|---|---|---|---|---|---|---|---|
| F | 16 | M0 | RR | 9872/3 | Negative | 46 XX | CD19 | 1.02 | 5.18 | D (1m) |
| F | 4 | RR | Negative | 44∼47XX,-2,+3,+4,+5,+der(5)t(5;?)(q33.3;?),+6, -8,-9,add(11) (q23.3),-14,-15,-18,+4mar [cp5] 46XX,add(11)(q23.3) [15] | CD2, CD7 | 1.07 | 8.18 | A (14m) | ||
| F | 8 | M1 | RR | 9873/3 | Negative | 46XX | CD7 | 1.00 | 12.66 | R (11m) |
| F | 7 | M2 | RR x t(8;21) | 9896/3 | t(8;21)(q22;q22) RUNX1(21q22.3) RUNX1T1(8q22) | 46XX, Inv3(q1;q26), t(9;22)(q34;q11.2) | — | No | 14.62 | A (37m) |
| F | 14 | RR x t(8;21) | t(8;21)(q22;q22)RUNX1(21q22.3) RUNX1T1(8q22) | 46XX t(8;21)(q22;q22) | CD38 | 1.04 | 8.62 | D (4m) | ||
| M | 17 | RR | 9874/3 | Negative | 48XY,+8,+21 | CD7 | 1.12 | 3.66 | A (15m) | |
| M | 11 | RR | Negative | 46XY | — | 1.09 | 7.18 | D (15d) | ||
| M | 6 | M3 | RR | 9866/3 | Negative | 46XY | — | 1.16 | 16.06 | R (6m) |
| F | 7 | M4 | HR x MLL | 9897/3 | Negative | 46XX t(4;11)(p14;q23.3) MLL | — | 1.00 | 2.35 | D (36m) |
| F | 6 | HR x MLL | t(11;19)(q23;p13.3) MLL(11q23) MLLT1 (19p13.3) | 46 XX | — | 0.99 | 5.75 | R (6m) | ||
| F | 10 | RR x inv(16) | 9871/3 | Negative | 46XX,add(5)(q35),inv(16)(p13;q22), -17,+mar [3] 46XX [17] | (Eos) | 1.01 | 62.21 | R (7m) | |
| M | 1 | M6b | RR | 9840/3 | Negative | 48XY, del(3)(p23p21),+6, t(6:22)(q21;q13),del(13)(q13q22),+21 [3] 48XYdel(13)(q13q22)+21 [6] 46 XY [11] | — | 1.08 | 6.38 | D (3m) |
| M | 14 | M7 | HR | 9910/3 | Negative | 47XY,+8 [5], 46XY [12] | — | 1.01 | 0.93 | D (20m) |
Thirteen of 81 cases (81%). WHO ICD-O-3, World Health Organization, International Classification of Diseases-Oncology-Version 3; Ab, aberrant; leu, leukocytes; PB, peripheral blood; M, male; F, female; RR, regular risk, HR, high risk; A, abandonment; R, remission; D, death; d, day; m, months; y, years.
Three cases of acute leukemia of mixed phenotype ALMP were observed accounting for 3.7% of AL (3/81; ICD-O-3 subgroup; 9805/3). Two were line B and monocytic; one had hypodiploidy (DNA index 0.8) and hyperleukocytosis (> 100,000 / μl) and is in induction to remission. The other patient had the t (9; 22) (q34; q13) translocation (WHO 9806/3) and abandoned treatment after one month of diagnosis. The third patient had B and M (undefined) phenotype and still is in remission after 58 months of diagnosis. All three are true bilinear cases (two populations of characterizable blasts), with no biphenotypic expression in a cell line or aberrant expression according to EGIL criteria.
3.2.4Acute lymphoblastic T leukemiaThe T-ALL group included three cases (3/81, subgroup ICD-O-3, 9837/3. CD45-, CD34-, nTdT+, CD1a+/-, CD2+, cCD3+, CD3+, CD4+, CD5+, CD7+, CD8+, TCRα / β+/-, TCRγ / δ+/ -). The other case was not subclassified. The three cases were enrolled with hyperleukocytosis (> 100,000 / μl). Hypodiploidy was identified in one of them and was accompanied by mediastinal tumor; in another, the STIL (1p32) alteration TAL1 (T-cell Acute Leukemia-1) was identified. A patient with more than 400 000 cells/μl in peripheral blood did not accept treatment and was discharged. The remaining two are still in remission 6 and 27 months after diagnosis, respectively.
3.2.5NK cell lymphoproliferative disorder (NK LPD)A case was identified with (1/81; ICD-O-3 9831/3) of NK LPD (CD45int, CD34+, CD10-, CD19-, CD13-, MPO-, CD2-, CD3-, CD4-, CD5-, CD8-, CD56 / 16+, CD58+with an aberrant expression of CD7) affecting a 2-year-old child, who evolved for 2 months with fever, severe joint and bone pain, pallor and hemorrhagic manifestations, without visceromegaly, peripheral pancytopenia, and t(11; 22) (p11.2; q12.3) translocation. The family applied for voluntary discharge after one month of admission.
3.3EthnicityAll the patients reported were native to the state of Chiapas, of which 60.5% (49/81) belonged to indigenous families, of which 55% (27) lived in rural areas of high or very high marginalization. The rest belonged to families with a varying degree of miscegenation located on the periphery of urban and suburban areas, as reported in their socioeconomic study.
4DiscussionThe results of 81 cases studied in detail from different points of view (morphological, immunophenotypic, and genotypic), with information on their evolution and short-term destination, allow to recognizing the types and diversity of AL that has been observed in the patients treated in the HEP and provide essential information regarding the inherent peculiarities of the disease in the attended population.
According to the primary objective of this work, and considering only the leukemic cell line, it is clear that native pediatric populations of Chiapas treated in the HEP showed a similar pattern to that seen in other parts of the country,10,11 despite their relative isolation and high degree of consanguinity. In most cases, B-lymphocytes were affected, followed by myeloid cells, T-lymphocytes and, isolated cases of phenotypic and other varieties, such as NK (Table 1).
Within B-leukemias, pro-B leukemias occurring in the first year of life (infant leukemia) were associated with MLL gene abnormalities and with frequent aberrant CD15 expression, as reported elsewhere. These leukemias exhibit aggressive behavior and are reported to have a poor prognosis.12–14 These facts suggest that pro-B leukemia is a particular disease produced by genetic alterations during gestation patient in an early lymphomonocytic precursor, and therefore, different from pre-B leukemia acquired at other ages.12,13 The 3.3% (2/61) of the B-ALL found in this population is comparable to the 1.2% (3/251) B-leukemia reported by Bekker-Méndez et al. in Mexican children (mostly mestizos) treated in Mexico City.15 Reports in other countries and other Mexican centers widely vary (from 0 to 65.4%). Due to its nature and poor prognosis, the approach of these cases could include early bone marrow transplantation, more aggressive treatments and strategies derived from the knowledge of the intimate molecular mechanisms, as a consequence of MLL gene alterations, such as the use of TAL1 inhibitors (a methyltransferase that interacts with transcriptional proteins associated with the MLL gene, inhibiting apoptosis of malignant cells), among others.16
Unlike the previous subgroup where the MLL gene alteration is determinant, in pre-B leukemias, quantitative and qualitative genetic alterations may or may not be present. However, when present, they give rise to specific subgroups with specific clinical characteristics. In this series, 36% of the B-ALL cases (22/61) presented them. Ten of the 22 (16.3% of B-ALL) consisted in translocations with or without karyotype alterations. This percentage is similar to the 17.7% reported by Bekker-Méndez et al.15 The most frequent translocation (6.6% B-AL) was t (1; 19) (q23; p13) (ICD-O-3; 9818/3), which fuses the PBX1 gene of chromosome 1 with the TCF3 gene (E2A) of chromosome 19, leading to a nuclear transcription protein 5′E2A / 3′PBX1, that constitutively activates transcription of genes regulated by the PBX family.17 Even when the presence of this translocation was originally associated with poor prognosis, it is now considered as a favorable prognosis marker due to current treatments.18,19 This perspective coincides with the evolution of the cases presented since the four cases with this translocation were still in remission five months to 1 year after the diagnosis, one of them despite having hypodiploidy and the other despite having extensive extramedullary disease. In the series of Bekker-Mendez et al. it was presented in 7.1% of B-ALL cases, and 19 of 20 patients were still alive after one year of diagnosis.15
The second most frequent translocation in this series was t (12; 21) (p13; q22) (ICD-O-3; 9814/3), which results in the fusion of the TEL (ETV6) genes of chromosome 12 and the AML1 on chromosome 21, with the subsequent production of the chimeric protein TEL / AML1 with has transcription activating functions of several hematopoiesis genes.20 Three patients presented it (5.0%) and, contrary to what was found in Western literature where it is reported as with good prognosis,21,22 all three cases, although they quickly achieved remission, died in relapse 1-4 years after diagnosis. This result differs from that observed by Bekker-Méndez et al. since, in this series, it was present in 7.4% of B- ALL, and only one patient (1/21) died in the first year. If this difference were confirmed with the analysis of more cases, an important local peculiarity would be established.
Three cases were observed with each translocation: t (1; 9) (q24; q34), t (1; 5) (q21; p15.3), and t (2; 3) (q21.3; p22). The first has been previously reported in ALL;23 it fuses the gene RCSD1 in chromosome 1, which encodes the CapZIP protein (related to the formation of the cellular cytoskeleton), with the ABL1 gene of chromosome 9, which encodes a specific tyrosine kinase. However, there are insufficient data in the literature regarding their clinical behavior. The patient with this translocation died ten months after diagnosis. The second translocation, t (1; 5) (q21; p15.3), has not been reported in leukemias. Genetic alterations were detected in a multi-treated 4-year-old female patient, who died ten months after diagnosis. The third case, who is in remission one year after diagnosis also presented hyperdiploidy. No reports of this translocation were found in the literature.
The absence of cases with the t (9:22) (q34; q11) in this subgroup is remarkable, as it is one of the reported changes in other domestic and foreign series.15,24,25 This translocation defines ALL cases with positive Philadelphia chromosome, who are, therefore, candidates for adjuvant treatment with currently available inhibitors of tyrosine kinase for treating classical chronic myeloid leukemia. Even with this resource, these cases have a poor prognosis. Thus, the translocation t (9; 22) has been incorporated as a criterion for group allocation to the VHR by the Children's Oncology Group (COG).22
Besides translocations, there are karyotype alterations; one of the most remarkable is the nonspecific hyperdiploidy without concurrent translocation corresponding to WHO ICD-O-3 group; 9815/3. It was present in 8 of 61 cases of B-ALL (13.1%), which is consistent with available international data.26 Results of this study suggest that hyperdiploidy is a key factor for favorable evolution in the absence of a specific translocation. Despite the initial classification risk by age, white blood cell count, and degree of infiltration of malignant cells, eight patients with hyperdiploidy were in remission four months to 1 year after diagnosis. Four additional cases presented various karyotype alterations: two were in remission, one died, and one was transferred to another institution (Table 4).
The subgroup of pre-B leukemias without genetic alterations is, as in other series, the largest group. These cases have behaved as in other centers: 20% died (1 month to 4 years from diagnosis), 11.4% discontinued treatment, and 68.5% remains in remission (2 to 19 months after diagnosis).
The two cases identified as mature B-leukemia are now considered a variant of Burkitt lymphoma (WHO 9826/3). Usually, they show c-Myc alterations secondary to t (8; 14) (q24; q32), determining the constitutive expression of Myc.8 The treatment of these cases as Burkitt lymphoma has radically changed its prognosis; a combination of chemo and immunotherapy is currently recommended.27 Cases in this series were not studied for c-Myc nor these translocations identified. They were treated according to the national protocol for leukemias, and their evolution has been favorable.
The relative frequency of AML observed in this study is similar to what has been observed throughout the country11,28 and the Hispanic population abroad,29 suggesting a connection with this ethnic variety. Classification into subgroups atomizes cases as the WHO includes at least 20 subgroups or AML variants associated or not with specific translocations.9 The distribution of these cases in the main FAB-WHO subgroups is somewhat homogeneous except for two M0 and two M2 cases with the t (8; 21), since the other subgroups provided a case for each of the observed subgroups (Table 5). In adults, it has been possible to establish three prognostic groups related to specific translocations.30 In pediatric patients, a greater diversity of opinions that generally incorporate the initial response to treatment exist.31
The scarcity of cases in this series does not allow a meaningful assessment of their behavior. Overall, regardless of their subclass, the evolution of cases with AML has been poor. Only with the study of more cases, it may be possible to establish an idea of the evolution of the different varieties and its comparison with international information.31
Acute leukemias with mixed phenotype (MPAL; WHO 9805-9809 / 3),8 which were previously known as bilinear biphenotypic, constituted 2% to 5% of leukemias, as in other countries.32 Three cases presented were BM (monocytic), but TM and BT can also occur. No specific national protocol for the treatment of this variety exists and, in general, there are no behavioral differences from those of a single cell line.
The T-ALL presented with rates comparable to the MPAL, in contrast to what is observed in Eastern countries, where it constitutes 15% of the AL. Classically, they are considered to have a poor prognosis, as they often are accompanied by hyperleukocytosis of >100 000 blasts / μl, mediastinal mass, tumor lysis, CNS infiltration, and secondary renal complications. However, with current treatments, a 5-year survival in over 60% of the cases has been achieved.33 The evolution of the reported cases has been very favorable, with good responses despite hypodiploidy and extensive disease.
To the extent of the immunological and molecular advances in the knowledge of leukemias, and its application to the identification, classification and sub-classification, clinical sub settings within the same affected cell line are established each with different behavior and prognosis characteristics. The existence of these subgroups raises the possibility of considering AL as a real syndrome, caused by several different molecular diseases. However, given the relatively low frequency of molecular distinctive punctual alterations (e.g., translocations ≈17%), it is also possible that AL is a single disease, which molecular basis remains hidden, and that the identified subgroups represent genetic and epigenetic alterations secondary to a primary common alleged alteration.
Native pediatric populations of Chiapas served by the HEP showed a ratio of primary leukemias similar to that seen in other parts of the country, despite their relative isolation and a high degree of consanguinity.
Other similar findings were the frequency, molecular alterations, and poor outcome of infant B-cell leukemia—usually with fatal outcomes—and the diversity of myeloid leukemias. Additionally, the good evolution of cases with translocation t (1; 19) (q23; p13) in B-cell leukemias and the favorable nature of hyperdiploidy in all cases.
Compared with national results, specific differences in this series also were observed: the poor outcome of B-cell leukemias with t (12; 21) (p13; q22) translocation (TEL / AML1) compared with the good prognosis in other places; the absence of the t (9:22) (q34; q11) translocation; no cases of trisomy 21, although the syndrome is relatively common in the local population; translocations not previously reported in leukemia, as t (1; 5) (q21; p15.3) and t (2; 3) (q21.3; p22); the good evolution of cases with leukemia / Burkitt lymphoma treated with the National Protocol for leukemias; the detection of a complex bilinear and myeloid case with unique behavior (e.g. M7 with subacute evolution); and the good outcome of T leukemias despite extreme hyperleukocytosis and hypodiploidy.
Probably, the near future, studying the entire genome of each patient will increase local differences. Consequently, medical research should continue with the comparative analysis of cases and their local particularities.26,34
This study was intended to serve as initial support for further analysis aimed at consolidating or rectifying these findings, with the ultimate goal of personalizing treatments for leukemia using both clinical and cytopathological characteristics and with risk assessment supported by objective local data.35
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
FundingThis study was made with resources from the Hospital de Especialidades Pediátricas Chiapas, Mexico; the Foundation Gonzalo Río Arronte and the Foundation Casa de la Amistad para niños con cancer IAP.
Conflict of interestThe authors declare no conflicts of interest.
The authors wish to express their gratitude to M. Sc. Joaquín Octavio Docelis Burguete and the engineer Ronald Gómez Martínez for their invaluable computer and biostatistical support, and to the Foundations Gonzalo Río Arronte and Casa de la amistad para niños Cancer IAP for the donation of the flow cytometry equipment and the reagents that made it possible to carry out this research.
Please cite this article as: Lepe-Zúñiga JL, Jerónimo-López FJ, Hernández-Orantes JG. Características citopatológicas de la leucemia aguda en un hospital de tercer nivel. Bol Med Hosp Infant Mex. 2017;74:122–133.