






1. Summary of the clinical history (A-11-23)
We present the case of a 5-year-old male patient with fever, abdominal pain and icterus.
1.1. Family medical history
The patient’s mother is an apparently healthy 27-year-old female with a primary school education. She is Catholic, denies substance abuse, and lives with a partner in a free union. The patient’s father is an apparently healthy 35-year-old male who is a mason. He is also Catholic. There is a family history of diabetes mellitus and liver disease.
1.2. Social history
The family is originally from and resides in the DF (Federal District). They own a home with basic services. The family lives with a pet dog.
Nutrition. The patient was integrated into the family diet with dietary indications by gastro-nutrition.
Psychomotor development. Psychomotor development was age appropriate.
Immunizations. Vaccinations were complete (in transplants, missing vaccinations are reported so they can be listed).
1.3. Perinatal and pathological history
The patient was the product of a second pregnancy with adequate prenatal care, normal ultrasonography, and intake of multivitamins. Pregnancy was of normal evolution and the patient was born at term, weighing 2700 g. Height and Apgar score are unknown. There were no perinatal complications. Patient was under treatment of the Hospital Infantil de México Federico Gómez (HIMFG) from January 2007 due to chronic liver disease with history of icterus since the first week of life.
July 4, 2007 (Pathology). A diagnosis of partial bile flow obstruction was made.
September 3, 2008. The patient’s condition was treated as autoimmune hepatitis and steroids were initiated. He later presented with elevated transaminases despite treatment. Steroids were slowly decreased and stopped. IgG was elevated, LKM (antimicrosomal antibodies type I of the liver and kidney) 2+, ANA (antinuclear antibodies) 1:80 fine mottling, antiribosome 9.4.
June 25, 2009. Treatment was initiated with prednisone for 2 months. In August the dose of steroids was decreased until it was terminated. There was no improvement in the aminotransferase levels.
September 8, 2010. A liver biopsy was performed, which was suggestive of a diagnosis of liver fibrosis.
1.4. Current illness
The patient presented with fever of 72 h evolution of sudden onset, two peaks quantified at 38°C with a 35-min duration. He had progressive, diffuse, moderately intense abdominal pain of 48 h evolution unrelated to food ingestion. He had sudden progressive onset of decreased consistency bowel movements of 24 h evolution on six occasions that were watery, greenish, foul smelling, and without mucous or blood. He had sudden, progressive onset of cephalocaudal icterus of 48 h evolution. He presented with alteration in the sleep-wake cycle of 24-h evolution. The mother reported lack of sleep during the night.
Physical examination revealed the patient to have a yellowish tinge with dry mucosa and hyporeflexic isocoric pupils. Abdomen was soft, distended, depressible without peritoneal irritation and globulous due to ascites. Peristalsis was not audible and there was hepatosplenomegaly and collateral venous network. Extremities were hypotrophic and capillary filling was 2”, with good color and temperature. Neurologically, the patient was irritable with Glasgow scale of 8 and without data of focalization or paresis.
Laboratory findings. Laboratory findings were as follows: leukocytes 9900, N 58%, L 15%, B 22%, Hgb 9.8, Hct 29, PLT 49,000, FA 558, BUN 36, calcium 7.7. Arterial blood gases were pH 7.31, pCO2 68.5, Sat 90.6%, HCO3 13.8, ammonia 233, PT 18.4, INR 1.45, PTT 33.
Admitting diagnosis. Diagnoses upon admission were as follows: severe acute malnutrition, cryptogenic liver cirrhosis, grade III hepatic encephalopathy, normochromic normocytic anemia, hyperuricemia, thrombocytopenia, septic shock, and probable cholangitis.
Evolution. Delayed capillary refill was reported up to 4” associated with deterioration of the neurological status (Glasgow of 8), for which reason phase III ventilation was begun. Subsequently, hypotension and tachycardia were reported and required crystalloid bolus. There was no satisfactory response. Dobutamine was begun and the Infectious Disease Service was consulted due to probable abdominal sepsis. The patient required empiric antibiotic coverage with ampicillin, cefotaxime and metronidazole for abdominal sepsis.
The patient was also evaluated by the Transplant Service and Pediatric Intensive Care Unit. HIV study and application of varicella vaccine were pending so as to be listed with the CENATRA (National Transplant Center).
He had anuria of 3-h evolution with high risk for multiple organ failure as well as hematochezia. It was decided to transfuse with red blood concentrate, fresh frozen plasma and platelets. Lower GI bleeding continued. There were up to 300 mL quantified. Four units of platelet concentrates, 225 mL of red blood concentrate and 150 mL of fresh frozen plasma were administered. Due to bleeding, the patient was evaluated by the Department of Surgery who suggested continuation of management based on fluids and transfusion. The patient continued to bleed and management with infusion octreotide was started. He had hypertension and bradycardia so the dose was decreased to 0.5 μg/kg. He was oxygen dependent with nasal cannula at 0.5 l/min. During the course of the day he had hematemesis (50 mL); therefore, a nasogastric catheter with bypass was placed and gastric lavage was done. He subsequently had hematochezia (700 ml/4h). Hgb was 8 g/dl, PT 19.6 and PTT. 28.8. He was again classified as NPO and transfusion with blood derivates was reinitiated. He had hypotension and decrease in urination (0.94 ml/kg/h). Octreotide infusion was increased to 1 μg/kg/h.
May 4, 2011. The patient was hemodynamically stable without bleeding and was oxygen dependent via nasal cannula. Hemoglobin was 0.3; therefore, it was decided to reinitiate oral feeding.
May 5, 2011. In a joint meeting with the Departments of Gastroenterology and Nutrition it was decided to perform sclerotherapy for grade III/IV varices due to the high risk of bleeding. Follow-up blood work reported Hgb 8.8; the prior number was 9.4.
May 6, 2011. Ligation of the varices and sclerotherapy was performed. On endoscopy the esophagus was observed with an incipient cord on the middle third, and at the distal third there were four varicose cords grade III-IV appreciated as well as evidence of a prior sclerosis. There was no active bleeding in the stomach. Next, the varices were ligated, which was done on two varicose cords. Subsequently, sclerotherapy was carried out on three varicose cords with polidocanol 1.5%. Octreotide was suspended. He had hypertension secondary to hypervolemia and a loop diuretic was administered.
May 10, 2011. The patient had two bowel movements with hematochezia and poor tolerance to oral intake. Hemoglobin was 9.8 and platelets 55,000. The patient was transfused with platelet concentrate and fresh frozen plasma. Octreotide was reinitiated. He also presented irritability and so ammonia was indicated.
May 13, 2011. The patient presented a sudden event of respiratory deterioration, data of encephalopathy and arterial saturation of 60%. Chest x-rays demonstrated bilateral macronodular infiltrate and he had gingival bleeding and epistaxis. Neurologically, he presented deterioration with alteration of the sleep/wake cycle, irritability and Glasgow scale 11; therefore, it was decided to intubate him. Subsequently, he suffered cardiorespiratory arrest; therefore, amines were given and advanced resuscitation was performed. During the event he presented massive hemorrhage of the GI tract and lung. Neurological status was Glasgow 3. PT 19, PTT 35, fibrinogen 197, Hgb 13, platelets 133,000, AST 98, ALT 35, IB 22, DB17. Two hours later he had another cardiorespiratory arrest of 9-min duration. The serious status of the patient was reported to the parents as well as the poor short-term prognosis. The parents decided that no advanced resuscitation maneuvers should be performed.
Discharge diagnoses
Discharge diagnoses were as follows:
— congenital cholangitic hepatic fibrosis
— portal hypertension
— grade III-IV esophageal varices in the distal third, large fundal varices and hypertensive gastropathy
— worsened chronic malnutrition of moderate intensity
— grade III hepatic encephalopathy
— controlled septic shock
— controlled pre-renal acute renal failure
— probable cholangitis
— massive pulmonary hemorrhage
2. Case presentation
2.1. Coordinator (Dr. Patricio Acosta Rodríguez)
The case presented is that of a 5-year-old male with multiple medical history of importance, among which is that of direct family members with liver and gallbladder pathology. He presented neonatal cholestasis, which was apparently resolved during the second month of life without factors that could be considered to be a direct cause.
Based on the signs and symptoms of the clinical history, the following syndrome diagnoses were made:
1. Cholestatic syndrome—on the basis of icterus from the first week of life and associated with direct bilirubin levels >1 mg/dl, with the total <5; not associated with acholia or hypocholia
2. Portal hypertension syndrome—manifested by ascites, collateral venous network and splenomegaly as well as esophageal and fundal varices according to the endoscopic findings reported.
3. Systemic inflammatory response syndrome—according to the presence of fever, hypotension and bandemia.
4. Acute diarrheal syndrome with dehydration—according to the presence of six bowel movements decreased in consistency of 24-h evolution associated with dryness of the mucosa
5. GI hemorrhagic syndrome—manifested by repeated epistaxis, hematuria, gingivorrhagia, hematemesis and melena
6. Brain syndrome—based on alteration of the sleep/ wake cycle, irritability as well as decreased level of consciousness without focal data
7. Acute respiratory insufficiency syndrome—based on tachypnea of sudden start, decreased oxemia and paroxysmal bilateral infiltrate
Based on the previously mentioned syndromatic diagnoses the following nosological diagnoses were made:
1. Chronic liver disease due to persistent elevation of aminotrasferases for a period >6 months, associated with cholestasis and elevation of gamma-glutamyl transpeptidase
2. Liver failure due to presenting changes such as coagulopathy, hyperammonemia, hypoglycemia, and hypoalbuminemia added to the hepatic encephalopathy described
3. Septic shock–based on the data of systemic inflammatory response with gastrointestinal and pulmonary foci, refractory to volume that required administration of vasoactive amines
4. Hypovolemic shock–due to presenting gingival, nasal, lower and upper GI tract bleeding associated with coagulopathy with hemodynamic compromise, added to massive pulmonary hemorrhage as a consequence of associated active bleeding to the sudden respiratory deterioration presented by the patient
This is a preschool-age child with baseline chronic liver disease without clear etiology. At the time of his referral to this institute, the patient had a very advanced stage of hepatic impairment evidenced by clinical data and supported by the biopsy that demonstrated biliary cirrhosis at 1 year 6 months of age. It is unknown if there was inflammatory infiltrate.
As the cirrhosis advanced, involvement of the liver architecture caused changes in the vasculature as well as the biliary structure. With it, the liver damage was perpetuated even when the initial problem ceased.
The fact that the patient had biliary cirrhosis from the first year of age implies extensive fibrotic liver damage commonly associated with an obstructive pattern and marked cholestasis. The fact of presenting pigmented bowel movements as well as the visibility of the gallbladder on echography does not rule out the possibility of an anatomic etiology, but it is considerably remote.
It is important to note that the patient should have been studied from the second week of life when he presented with icterus. Having documented neonatal cholestasis and without giving importance to the presence or absence of pigment in the bowel movements, a complete diagnostic approach should have been provided. Given the complexity of the approach and diagnostic considerations, it is important to evaluate the available clues.
Liver cirrhosis can be a result of multiple causes. In the case of this patient, visceromegaly makes one suspect disease by deposits, particularly liposomal deposit. Similarly, one of the predominant causes of biliary cirrhosis is cystic fibrosis, which was apparently ruled out.
Upon continuing with the diagnostic considerations, it was observed that in this patient the infectious causes became highlighted on the face of the history of neonatal cholestasis. In the summary of the clinical history no evidence was found of having ruled out cytomegalovirus, Epstein Barr, toxoplasmosis, rubella, HIV, which occasionally have liver involvement. Also, the possibility of it being a recurrent viral infection was not ruled out. In fact, the patient should have been immunized for hepatitis A, hepatitis B and chickenpox due to the risk of acquiring new infections, so as to avoid greater liver damage.
Clinically important toxins are another entity to rule out in this type of patients due to the cultural practices of consumption of teas, fungi and other substances that can affect the liver to such a degree as to cause fulminant liver failure that was not present in the patient. Another diagnostic possibility is hepatic veno-occlusive disease; however, this is not supported by the histopathological findings reported in the clinical files.
An important point is to suspect autoimmune hepatitis in the patient given that it is a treatable cause of chronic liver disease. This is mandatory because of the characteristics already described as well as the presence of anti-microsomal, renal-hepatic type one and antinuclear antibodies which, at a point in time, required steroid treatment for 6 months. However, according to the established diagnostic criteria by the international study group of autoimmune hepatitis (modified and simplified in 2008), one must include compatible liver histology, selective hypergammaglobulinemia, positivity of antibodies and exclusion of viral infections and Wilson’s disease. The available information is insufficient to integrate or rule out this diagnosis. In the presence of a patient with the aforementioned history who clinically manifests with portal hypertension and biochemically with discrete elevation of the aminotransferases, minimum cholestasis and without involvement in the hepatic synthesis, it would appear not to agree with the diagnosis of biliary cirrhosis reported in the liver biopsy. However, there is a disorder that combines with this presentation: congenital hepatic fibrosis in which the more fibrotic forms are histologically confused with biliary cirrhosis. Usually, periportal fibrosis and irregular proliferation of the bile ducts tend to be observed, thereby causing venous obliteration. Congenital liver fibrosis is commonly associated with kidney damage, principally manifested by polycystic kidney and nephroptosis. In this patient, no disorder of this type was found, with the exception of hematuria. Although this disease normally has a benign course, malformation of the duct plate causes multiple degrees of portal hypertension in up to 70% of the patients affected and it becomes the main reason for patient morbidity and mortality.1-4
The usual age of presentation is adolescence; however, case series reported worldwide show a wide range of phenotypes with an autosomal recessive inheritance and predominantly liver involvement that could be present from the neonatal period until adolescence and adulthood. Based on what has been described, it is important to emphasize that the study of chronic liver diseases according to its degree of complexity should be done in an orderly and systematized manner to achieve a timely diagnosis. Special attention should be paid to the clinical presentation because, despite the presentation, in a significant number of patients its etiology is not able to be defined.5-8
The main complication presented by the patient was upper GI bleeding secondary to intrahepatic portal hypertension due to periportal fibrosis and sinusoidal compression. He also had progression in prehepatic venous dilation.
During the patient’s bleeding events, it was not reported whether a timely endoscopic intervention was done, i.e., within the first 24 h of its initiaion and once the pertinent fluid resuscitation has been done. No treatment of the gastric varices was done. No treatment of any type was done for the gastric varices. Due to their complexity they represent an important therapeutic challenge because tissue adhesives can be used. In fact, exclusive treatment of esophageal varices tends to aggravate the previously existing fundal varices, causing increased bleeding risk.
With respect to transfusion of blood derivatives, contrary to common beliefs, administration of fresh frozen plasma is not recommended for patients with cirrhosis and upper GI bleeding with the purpose of correcting a coagulopathy because it has not demonstrated beneficial in the patient’s prognosis. It is also not indicated to be used prophylactically prior to endoscopy because it causes an increase in the plasma pressure and therefore in the portal pressure, increasing the risk of bleeding as occurred in this case. Administration of platelet concentrates is recommended only when there is a platelet count <30,000 and a recommended hemoglobin target between 7 and 8 g/dl.
During lower GI bleeding presented as massive hematochezia, it is currently recommended that an angio-tomography and scan be performed so as to localize the bleeding. If the site is not seen a diagnostic and therapeutic colonoscopy should be performed given that in up to 75% of the cases the bleeding site is identified and in up to 95% endoscopic hemostasis is achieved. Lower gastrointestinal tract bleeding presented as massive hematochezia. Angiography and scan were recommended in order to locate the bleeding site. For this reason, it is widely recommended that colonoscopy be performed during the first 24 h of active bleeding and after endoscopic intervention to use analogs of somatostatin or terlipressin for 3 to 5 days. The patient was not provided with the benefit of any of these diagnostic or therapeutic studies even though these treatments are not considered to be curative but palliative.
With respect to the primary prophylaxis of GI bleeding in patients with portal hypertension and biliary cirrhosis, use of nonselective beta blockers such as propranolol have little evidence demonstrated in children. However, in adults, studies demonstrated a decrease in the incidence of bleeding of up to 50%, a reason why its use is common in pediatric patients such as in the present case. Despite this, it has not been possible to evidence a significant difference between these treatment behaviors.
As secondary prophylaxis, ligation of the varices was performed. There is insufficient evidence on the use of propranolol to either justify or contraindicate its use. With regard to gastric varices, the use of cyanoacrylate (a tissue adhesive) has shown a level of effectiveness similar to that of the ligature, with a lower rate of bleeding recurrence. However, these are therapeutic challenges worldwide. This type of patient is considered to be an ideal candidate for the performance of postsystemic short circuits given that liver function is preserved. For this, a vascular bypass should have been considered, significantly decreasing morbidity and thereby patient mortality. In the present case the etiology of the fibrosis was unclear, and prior to carrying out a liver transplant, it is important that the site not be surgically manipulated. Another important complication presented by the patient was ascites, which increased due to the decrease in osmotic pressure and increase of the renin-angiotensin-aldosterone system, which conditions a leak to the third space, principally at the peritoneal level. This patient received a mixture of IV solutions with high amounts of sodium, which perpetuated or increased the ascites during hemodynamic decompensation. Treatment of the ascites passes on to a second level and should be taken
into account once the patient has been stabilized. Finally, after 5 years of experiencing repeated bleeding episodes, he presented with an infectious picture associated with hepatic encephalopathy, with prolonged times of coagulation and INR >1.5 despite an adequate administration of vitamin K. This is defined as acute liver failure in the context of a patient with chronic liver disease.
Within the management of hepatic encephalopathy, the use of lactulose was correct; however, it was initiated in the second week of hospitalization. In place of lactulose one can use antibiotics such as rifaximine that have little enteral absorption with similar effectiveness as lactulose and fewer side effects.
It is important that once hemodynamic stability has been obtained, an adequate caloric intake is ensured in order to avoid catabolism. It is recommended to begin with a dose of protein of 0.5 g/kg/day with slow increases until reaching 1.5 g/kg/day. The use of vegetable protein should be preferred because of its smaller quantity of methionine and aromatic amino acids. All of these measures coupled with appropriate blood sugar and electrolyte balance have proven to be an essential part of the treatment of hepatic encephalopathy.
Episodes of serious infection that the patient presented should not be surprising given that liver failure conditions a lower production of immunoglobulins and complement system factors, thus affecting the innate and adaptive immunity of the patient. Similarly, hypersplenism conditions a decrease in leukocyte count and function, leading to a secondary state of immunocompromise compounded by the multiple conditions previously mentioned according to the medical history.
A fundamental point to be made about this patient is the benefit that would have been presented if a liver transplant had been performed. It has been described that up to 7% of patients with congenital liver fibrosis who live beyond the neonatal period will require a transplant to resolve the complications of portal hypertension and recurrent cholangitis. Two points for reflection are the long period that elapsed between the first contact with the institution and the beginning of the transplant protocol. Even though it was known by the majority of the services involved, it was not able to be completed during the 35 months after initiation. The patient showed hemodynamic and ventilatory deterioration, both due to the infectious processes as well as the multiple bleeding episodes, progressing to multi-organ failure (Table 1).
Final diagnoses are as follows:
• Biliary cirrhosis probably secondary to congenital liver fibrosis, which caused portal hypertension, esophageal and fundal varices, and hypertensive gastropathy
• Upper and lower GI bleeding
• Acute liver failure in the context of chronic liver disease
• Short stature in relation with weight/age and weight/ height not evaluated for organomegaly and ascites
• Septic shock with intestinal and pulmonary foci
Immediate cause of death:
• Hypoxia due to respiratory insufficiency secondary to massive pulmonary bleeding
2.2. Imaging (Dr. Miguel Flores Armas)
Of the imaging studies performed, the most important are mentioned. On the imaging of April 21, 2010 there is presence of a cannula within the stomach; in the chest a mild increase of the bilateral interstitial radio-opacity and mild widening of the mediastinum were observed. CAT scan of the central nervous system was without relevant findings. Chest x-ray done on April 23 demonstrates increased bilateral perihilar radio-opacity most apparent on the left upper lobe. The thoraco-abdominal projection done on October 21 shows abdominal distention, changes in the gas distribution predominantly towards the central region and also an increase in liver volume, which surpasses the costal margin and is close to the ribs on the left side. On January 10, 2011 there was increase of the parahilar radio-opacity in the right lung without reaching the periphery. On April 23, 2011, chest x-ray showed increase of the radio-opacity of the left lung, asymmetry of the hemidiaphragm and widening of the left side of the mediastinum.
3. Discussion (Dr. Alejandro Hernández Plata)
As explained above, given the complexity of this case it was not easy to reach an etiological diagnosis of liver disease. Therefore, the matter would be to know if there was sufficient evidence to suspect autoimmune hepatitis. Currently, if the diagnosis of autoimmune hepatitis is suspected, the question would be the validity of treatment.
3.1. Gastroenterology (Dr. Rodrigo Vázquez)
Based on data from the clinical history and data of liver biopsy, diagnosis of autoimmune hepatitis was unlikely. Knowledge about how to diagnose and treat autoimmune hepatitis has been increasing, but in regard to being a cause of chronic liver disease it should be taken into account because it is feasible to treat autoimmune hepatitis and to see if there is clinical improvement. Although it is not a criterion, it may support the diagnosis. Based on the clinical history, liver damage corresponded to cirrhosis of the liver, but inflammatory activity was not demonstrated. The presence of antibody D was ruled out because a damaged liver can contain the same elevation. On this basis, this would not be autoimmune hepatitis. If there are clinical, laboratory and histological criteria that suggest the diagnosis, treatment can be initiated and the response evaluated.
3.2. Transplants (Dr. Alejandro Hernández Plata)
Part of the treatment for chronic liver disease is to treat the complications of portal hypertension such as esophageal varices. The following questions arise: When should an endoscopy be performed? How should the esophageal varices be treated? What is their follow up?
3.3. Thoracic surgery and endoscopy (Dr. Iván Rivas Rivera)
First we need to define what is primary prophylaxis and secondary prophylaxis. Primary prophylaxes are those pharmacological and endoscopic measures that are performed in the patient in whom esophageal varices have already been confirmed. The use of nonselective beta blockers in pediatric patients is still under discussion as opposed to adult patients in whom it has already been proven. As is often common, in the pediatric population there is a lack of randomized and controlled studies to recommend treatments. With regard to endoscopy, it has been concluded that variceal sclerosis in a patient who has not bled is not an appropriate treatment, but not so for ligature of the varices. This procedure has the limitation of the equipment that is used. To ligate a varix, one needs a special barrel which, because of its size, is limited to be used in patients weighing >13-15 kg. If it is possible to pass the endoscope with a barrel, it is feasible to perform prophylactic ligature of the varices. Secondary prophylaxis is done in a patient who has varices that have already bled. Pharmacological treatment and treatment based on sclerosis should be continued. It is important to rule out the presence of gastric varices prior to treatment of the esophageal varices. If there are large gastric varices at risk of bleeding and the esophageal varices are treated, there is a risk of increasing the flow to the gastric varices, causing bleeding. Treatment with cyanoacrylate in pediatric patients has not been completely validated. It is only recommended in certain patients, for example, those who do not have esophageal or gastric varices. In conclusion, one must have a very clear idea of the type of prophylaxis that a patient can be provided in order to determine if he/she is a candidate for sclerosis or for ligature of the varices.
4. Pathology (Dr. Mario Perezpeña Diazconti)
As has already been mentioned in the clinical history, the first contact with the patient was a percutaneous liver biopsy. A cylinder was obtained in which we were able to observe that the greater part of the biopsy corresponded to fibroconnective tissue and small hepatocyte nodules (Fig. 1A). With Masson trichrome staining, everything corresponding to fibrosis was observed in blue and small remnants of hepatocytes in red.
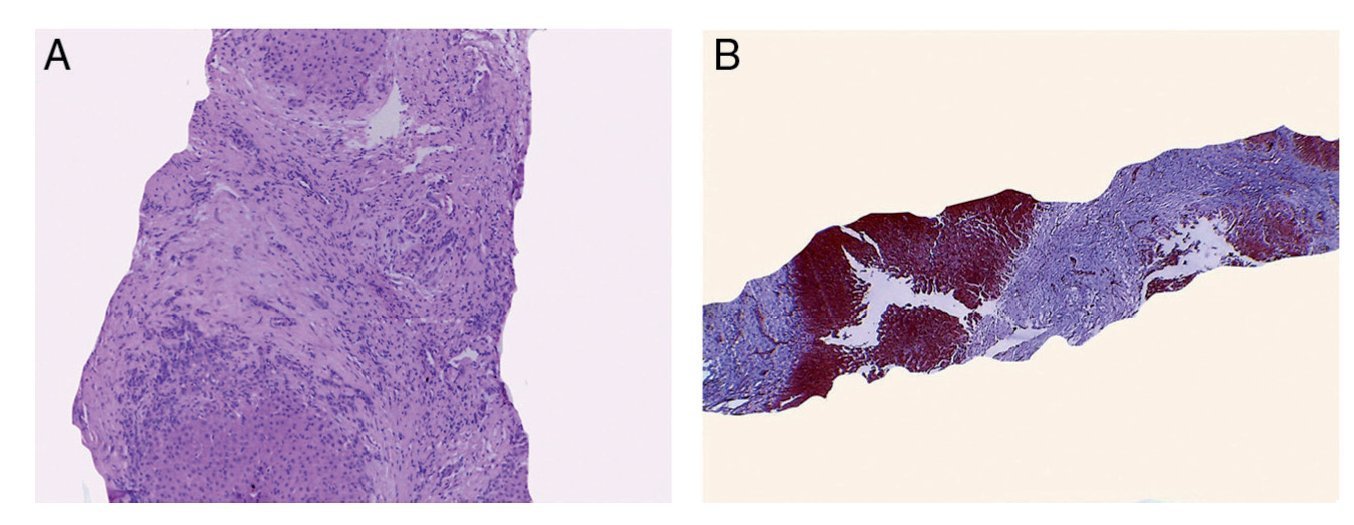
Figure 1 (A) Hepatic biopsy showing extensive fibrosis with small remaining nodules of hepatocytes without alterations. (B) Diagnosis was biliary cirrhosis secondary to partial obstruction of biliary flow.
Liver biopsy demonstrates better results when we are dealing with diffuse liver diseases as disease changes can be observed in any part of the biopsy. However, in particular, in the disease presented by the patient more damage was demonstrated in the subscapular region and in the left lobe of the liver. On the other hand, there are occasions during which it is very difficult to arrive at a diagnosis because the liver reacts in the same manner in the face of different stimuli. In this case, diagnosis of biliary cirrhosis secondary to partial obstruction of the biliary flow was done (Fig. 1B). One must not immediately consider bile duct atresia because there are many diseases that may present this pattern. With electron microscopy (EM), some metabolic and mitochondrial diseases were ruled out. EM report showed cholestatic changes and portal hypertension. At the time that the autopsy was done there was hematic fluid found in the two pleural cavities. A complete exploration was done to look for some type of obstruction, which was ruled out.
The liver parenchyma demonstrated abundant thin fibrous septa of whitish-yellow color. The thickness was greater below the Glisson capsule and formed small nodules, explaining why on the biopsy a large quantity of fibrosis was noted (Fig. 2). This disease affects the periphery of an organ with greater intensity and decreases toward the central part of the liver. As has already been mentioned, it is a case of congenital liver fibrosis. This disease is due to an alteration in a protein called fibrocysteine. The cytogenic alteration is a mutation in the short arm of chromosome 6 where the PKHD1 gene (of polycystic kidney and liver disease) is found. Dysfunction of this protein affects the bile ducts of the liver, the collecting tubules of the kidney and the pancreatic ducts (Fig. 3). Histologically, there is fibrosis in the portal spaces and formation of irregular, cystic ducts due to fusion of the ductal plate. The ductal plate is two layers of cells located around a portal vein during its fusion and remodelation. When there is lack of this protein, there is an increase in the number of bile ducts formed. Intrahepatic bile ducts, which are dilated, are tortuous and the epithelium is flattened. The portal veins also demonstrate changes; they are increased in number and are small.

Figure 2 Liver, biliary vesicle and extrahepatic biliary tracts that were explored and found permeable are observed. In addition, pancreas and duodenum are shown. In the liver parenchyma there are abundant thin fibrous septa of connective tissue that become thicker as they approach the periphery. The capsule is fibrous.
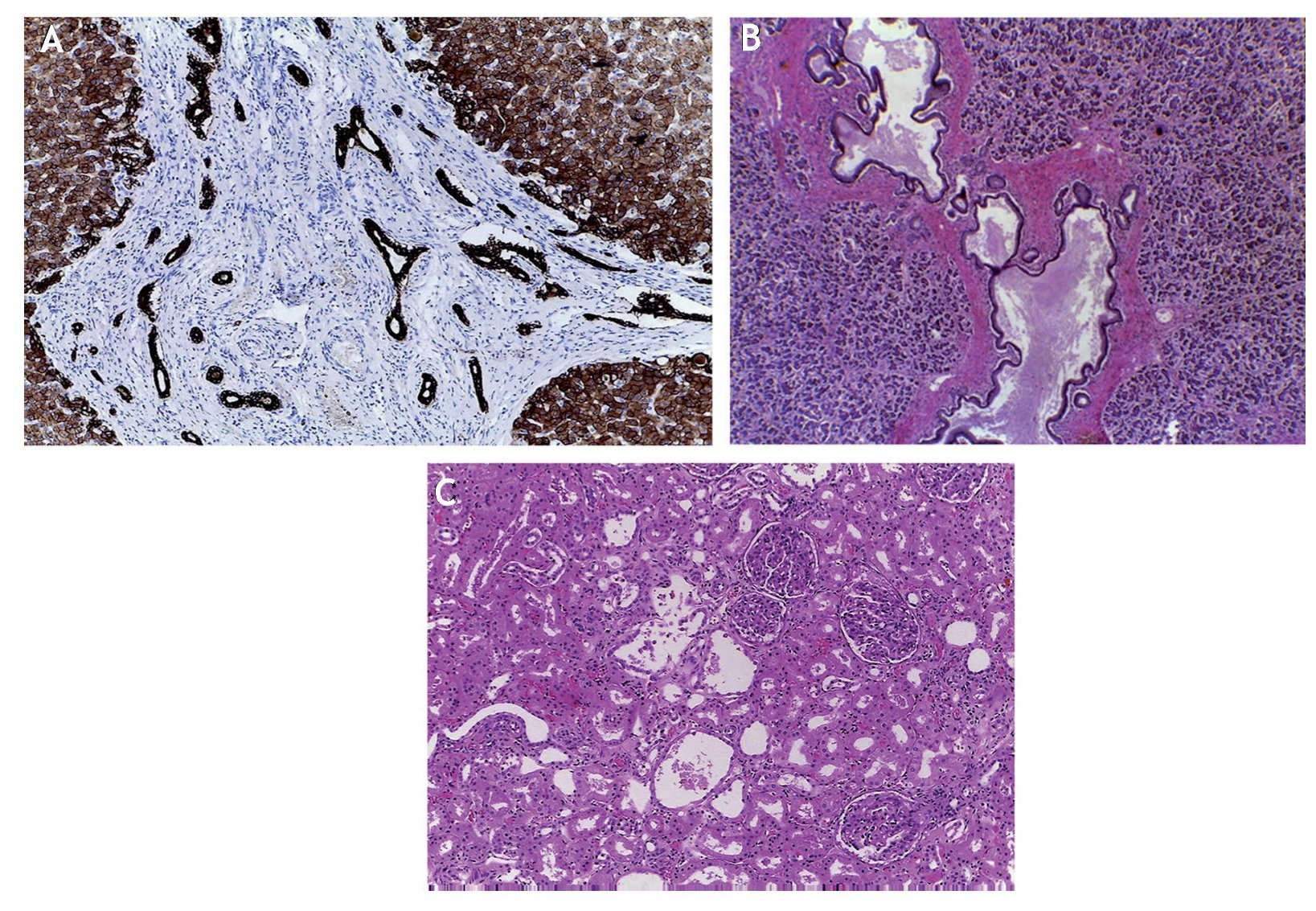
Figure 3 Characteristic damage is shown of diseases in (A) hepatic biliary ducts, (B) pancreas and (C) renal tubules.
In the pancreas, the ducts are dilated and have dense eosinophilic material in its lumen. Necrosis of the epithelial component and adipose tissue is also seen. The other organ affected is the kidney (Fig. 4). Both kidneys are increased in size and weight as has been described in autosomal recessive polycystic kidney disease. Histologically, small cysts originating in the collecting tubules covered with flattened cubic epithelium are observed and correspond to the terminal branches of the collecting tubules. In this disease, when the renal changes are more apparent, liver damage is less and vice-versa.
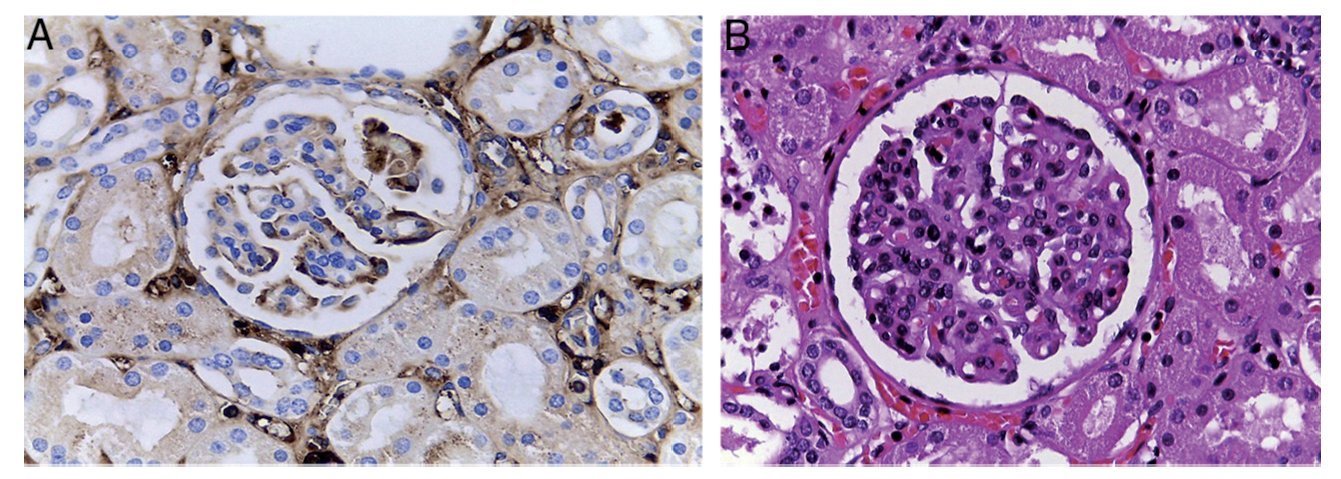
Figure 4 Renal damage due to cirrhosis.
Due to liver damage, the kidney experiences other changes such as focal glomerulonephritis, mesangial proliferation due to IgA deposits, liver cirrhosis and liver failure, and damage due to bile material in the kidney tubules. Also, because of the liver damage, in the esophagus and stomach there are dilated and congested varices associated with ulcerated areas in the esophagus. In these areas there is fibrin, cellular detritus and yeast, which could correspond to Candida sp. Thirty five mL of blood was found in the stomach.
The lungs were increased in size and weight with extensive areas of reddish-brown color. Histological cuts demonstrated extensive areas of pulmonary hemorrhage. There were no data of infection found. Focally there were hyaline membranes associated with alveolar damage. There was edema and dilated lymph vessels seen in the pleura. The spleen was increased in size and weight with chronic passive congestion.
In conclusion, this was a patient with congenital liver fibrosis in whom the renal and pancreatic damage was discretely present.
5. Final comments
5.1. Nephrology (Dr. Luis Velásquez Jones)
The majority of the studies in patients with chronic liver disease who developed a renal disorder have been done in adults. Very few studies have been carried out in the pediatric population. In adults, 50% of patients with cirrhosis have an IgA elevation. An IgA elevation was noted in this patient. It is known that adult patients with cirrhosis develop mesangial proliferation of the kidney. According to the immunofluorescence study, there are abundant IgA deposits found that correlate with the elevated serum levels of IgA observed in 90% of these patients. In this case the patient presented with hematuria and elevated IgA levels. It can be said with great certainty that he had an IgA nephropathy with creatinine elevation. This disease is uncommon although secondary IgA nephropathy has been reported in patients with biliary duct atresia. After renal transplant, in these patients the IgA nephropathy disappears.
Conflict of interest
The authors declare no conflict of interest of any nature.
Received 23 September 2014;
accepted 2 October 2014
* Corresponding author.
E-mail: mpdiazconti@gmail.com (M. Perezpeña-Diazconti).