



Developing a novel drug is a complex, risky, expensive and time-consuming venture. It is estimated that the conventional drug discovery process ending with a new medicine ready for the market can take up to 15 years and more than a billion USD. Fortunately, this scenario has recently changed with the arrival of new approaches. Many novel technologies and methodologies have been developed to increase the efficiency of the drug discovery process, and computational methodologies have become a crucial component of many drug discovery programs. From hit identification to lead optimization, techniques such as ligand- or structure-based virtual screening are widely used in many discovery efforts. It is the case for designing potential anticancer drugs and drug candidates, where these computational approaches have had a major impact over the years and have provided fruitful insights into the field of cancer. In this paper, we review the concept of rational design presenting some of the most representative examples of molecules identified by means of it. Key principles are illustrated through case studies including specifically successful achievements in the field of anticancer drug design to demonstrate that research advances, with the aid of in silico drug design, have the potential to create novel anticancer drugs.
El desarrollo de un nuevo fármaco es un proceso complejo y arriesgado que requiere una enorme cantidad de tiempo y dinero. Se estima que el proceso estándar para producir un nuevo fármaco, desde su descubrimiento hasta que acaba en el mercado, puede tardar hasta 15 años y tener un costo de mil millones de dólares (USD). Por fortuna, este escenario ha cambiado recientemente con la llegada de nuevas tecnologías y metodologías. Entre ellas, los métodos computacionales se han convertido en un componente determinante en muchos programas de descubrimiento de fármacos. En un esfuerzo por incrementar las posibilidades de encontrar nuevas moléculas con potencial farmacológico, se utilizan técnicas como el cribado virtual de quimiotecas construidas con base en ligandos o estructuras para la identificación de hits y hasta para la optimización de compuestos líder. En lo que respecta al diseño y descubrimiento de nuevos candidatos a fármacos contra el cáncer, estos enfoques tienen, a la fecha, un impacto importante y aportan nuevas posibilidades terapéuticas. En este artículo se revisa el concepto del diseño racional de moléculas con potencial farmacológico, ilustrando los principios clave con algunos de los ejemplos más representativos y exitosos de moléculas identificadas mediante estas aproximaciones. Se incluyen casos desarrollados en el campo del diseño de fármacos contra el cáncer con la finalidad de mostrar cómo, con la ayuda del diseño asistido por computadora, se pueden generar nuevos fármacos que den esperanza a millones de pacientes.
The Nobel Prize in Physiology or Medicine 1945 was awarded jointly to Ernst Boris Chain, Sir Howard Walter Florey and Sir Alexander Fleming for the mass production of penicillin discovered by the latter almost two decades before. At that time, the average life expectancy at birth in Mexico was about 45 years.1 Currently, life expectancy for newborns is around 75 years. This increase can be explained by many factors, including the significant amount of medication that physicians prescribe today to extend the life of a patient.
Most of the effects of medicines are based on the interaction between therapeutic chemical compounds (drugs) and proteins (targets), such as G-protein-coupled receptors, ion channels, proteases, kinases or nuclear hormone receptors, among others. Therefore, we might wonder how many new drugs are still to be discovered by researchers. The answer would be almost an ‘unlimited tm) quantity. The number of potential targets remains unclear. However, recent estimates claim that the number of current targets is over an order of magnitude lower than it could be.2,3 On the other hand, the number of organic molecules that can act as drugs is also a matter of debate. More than 100 million small chemical compounds have been already synthesized for their screening on specific targets in public and private laboratories.4 In addition, it is not only a matter of quantity but also of quality. Until recently, medicines have been discovered either as a result of unexpected investigations•by inspection of natural substances traditionally considered as therapeutic•or in extensive experimental blind screening studies.5 These drugs, although proven to be useful in the treatment of several pathologies, have significant downsides, like important side effects or low efficiency. As they were discovered, not designed, new drug generations have to overcome these difficulties.
The process of discovery and development of novel drugs is known to be time-consuming and expensive. On average, the standard process of discovery and development of drugs to marketing takes from 10 to 15 years. Furthermore, the average cost for research and development of each effective drug is estimated at $1.8 billion USD.6 In this context, it is not surprising that the development of new strategies over the last decades has emerged to make the processes much more rational and efficient using new supercomputers combined with the knowledge and experience of researchers. To this day, physicians, biologists, chemists, physicists, and computer scientists work, hand in hand, with the goal of offering to patients better and more selective drugs to improve their quality of life. Nevertheless, there is still a long way to go in the field of drug discovery, or should we say, drug design?
In this article, we first introduce the concept of computer-aided drug discovery and design with a summary of the leading computational techniques that include drug-repositioning approaches and lead optimization techniques. In the second section, we describe how these approaches have been successfully applied. In addition, we review the concept of rational design and present some of the most representative examples of molecules identified by its means. Key principles are illustrated through case studies exploring the field of anticancer drug design to demonstrate that research advances, with the aid of in silico drug design, have the potential to create novel anticancer drugs.
2Computer-aided drug discovery and designSince the advent of the X-ray diffraction to unveil the chemical composition and three-dimensional (3D) geometry of a small organic molecule in 1932,7 a large number of proteins have been solved either by X-ray or by nuclear magnetic resonance (NMR) spectroscopy and are available at open access protein databases (http://www.rcsb.org). This information allows researchers to understand and characterize many physiological processes based on interactions between proteins or between proteins and small molecules (ligands), as the case of the drug-target binding.
In 1962, Max Perutz and John Kendrew were awarded the Nobel Prize in Chemistry for the first solved high-resolution structure of protein (myoglobin). Since then, several other studies in crystallography determination of protein structure have been awarded the Nobel Prize8 until the recent Nobel Prize in Chemistry 2012, which was awarded jointly to Brian Kobilka and Robert Lefkowitz for the structural and functional studies on G-protein-coupled receptors (GPCRs).
With the chemical composition and 3D relative position of each atom in a target, the quest to find hit molecules that could potentially act as drugs has evolved considerably: from a blind screening process that hopes for finding molecular hits essentially by serendipity to an approach often called ‘rational tm) drug discovery and design5. In the 80's, this was the case of the first angiotensin-converting enzyme (ACE) inhibitor Capoten (captopril), the first drug optimized using structural information. In 1997, nelfinavir mesylate (Viracept)•an HIV protease inhibitor•was the first drug with a design completely driven by the structure of the target approved for the US market.9 These discoveries were only the beginning of a frantic career in search of novel, faster, and cheaper methodologies, and computational algorithms and techniques to develop and design new drugs. Moreover, to sample more compounds over the target (screening process) in less time and to acquire a priori key knowledge and expertise to design the library of chemical compounds for further screening in a more precise manner.
After solving protein structures at high resolution, the relevant revolution came when computational models based on simple physical laws were able to mimic the interactions between the organic molecules, atom by atom. Apart from the 3D structure of a molecule, the electrostatic charges and dipoles, the atoms tm) van der Waals radii, the parameters of covalent bonds, torsions, and dihedral angles were considered. Researchers today can approach real systems by using virtual or in silico experiments with the support of computational facilities, such as powerful workstations or supercomputers. This advancement has been the keystone, which has paved the way for a more rational approach to the query of efficient, selective and fewer side effects drugs, and at the same time making the process cheaper and less time-consuming.
At present, with these technologies, screening more compounds in less time at a lower cost is possible (virtual screening). The Computer-Aided Drug Discovery and Design (CADDD) era, where computer simulations of chemical systems have triggered the possibilities in this field, has allowed researchers to make in silico improvements. Among these advances, there are the resolution of 3D structures using computer models, the optimization and design of new compounds, and the understanding and characterization of the atomic mechanisms of previous drugs or natural substances. Furthermore, breaking the paradigm of orthosteric drugs (drugs binding to the target at the specific active site) to expand the search of therapeutic molecules to allosteric modulators and bitopic drugs.
The impact of these methodologies, as well as the science development in understanding and modeling the all-atom chemical and biochemical processes and reactions from a computational and physical perspective, has recently been recognized with the Nobel Prize in Chemistry (2013) to the physicists Martin Karplus and Michael Levitt together with the chemist Arieh Warshel.
Currently, many technologies have been developed to boost the efficiency of the drug discovery process. Since its emergence, CADDD has experienced a rapid increase in development, to which many different research groups around the world have made significant contributions. It has also emerged to harness various sources of information to facilitate the development of new drugs that modulate the behavior of therapeutically interesting protein targets, accelerating the early-stage pharmaceutical research. Rapid developments in CADDD technologies have provided an environment to expedite the drug discovery process by enabling huge libraries of compounds to be screened and synthesized in short time and at a very low cost. Today, CADDD is a widely used term to represent computational tools and sources for the storage, management, analysis and modeling of compounds10 used at almost every stage of a drug-discovery project, from lead discovery, optimization, target identification and validation, to even preclinical trials.11
3Ligand- and structure-based methodsEvidence of computational drug design success in the field of drug development is reflected in a significant number of new drug entities that are currently in clinical evaluation.
Computational drug design has emerged to harness different sources of information to facilitate the development of new drugs that modulate the behavior of therapeutically interesting protein targets. These computational approaches are classified mainly into two families: ligand- and structure-based methods.
Ligand-based methods use the existing knowledge of active compounds against the target to predict new chemical entities that present similar behavior.12 Given a single known active molecule, a library of molecules may be used to derive a pharmacophore model to define the minimum necessary structural characteristics a molecule must possess in order to bind to the target of interest. Comparison of the active molecule against the library is often performed via fingerprint-based similarity searching, where the molecules are represented as bit strings, indicating the presence/absence of predefined structural descriptors.13
In contrast, structure-based methods rely on targeting structural information to determine whether a new compound is likely to bind and interact with a receptor. One of the advantages of the structure-based drug design method is that no prior knowledge of active ligands is required.14 From a drug 3D structure it is possible to design new ligands that can elicit a therapeutic effect. Therefore, structure-based approaches contribute to the development of new drugs through the discovery and optimization of the initial lead compound.
Currently, the combination of ligand- and structure-based methods has become a common approach in virtual screening since it has been hypothesized that their integration can enhance the strengths and reduce the drawbacks of each method.
In this section, some of the most representative computational approaches used to design, optimize and develop a new drug are described. Although there are remarkable differences among them, they share a common goal: harvesting potential ligands or hits with the capability to bind to the target from an extensive database of generic small chemical compounds (Figure 1). To achieve this goal, many essential steps and decisions have to be made in order to eliminate from irrelevant compounds at the beginning, to end up with those that show better potential activity or have side effects and show interaction with other drugs. This process performed with the assistance of computational algorithms is called virtual screening.
 Standard in silico drug design cycle consists of docking, scoring and ranking initial hits based on their steric and electrostatic interactions with the target site, which is commonly referred to as virtual screening. Generally, in the absence of structural information of a receptor, and when one or more bioactive compounds are available, ligand-based virtual prescreening is utilized. This prescreening method is carried out by similarity search. The basic principle behind similarity searching is to screen databases for similar compounds with the backbone of the lead molecule. (B) In many situations, 2D similarity searches of databases are performed using chemical information from the first generation hits. (C) One alternative approach employs a ligand-based pharmacophore strategy that is often partnered with structure-based docking that uses a more stringent scoring matrix to determine the relative score made by matching two characters in a sequence alignment. This enhances the enrichment of initial hits and identifies the best compounds for computational evaluation, which are the second generation hits. (D) In the second phase, the molecular interactions between the target and the hits often identify ligand-based sites for optimizing these metrics for a unique molecular chemotype. (E) Computer algorithms, compounds or fragments of compounds from a database are positioned into a selected region of the structure (docking). These compounds are scored and ranked based on their steric and electrostatic interactions with the target site. (F) Structure determination of the target in complex with a promising lead from the first cycle reveals sites on the compound that can be optimized to increase potency.")
Workflow for hit identification: from data preparation to finding new leads. (A) Standard in silico drug design cycle consists of docking, scoring and ranking initial hits based on their steric and electrostatic interactions with the target site, which is commonly referred to as virtual screening. Generally, in the absence of structural information of a receptor, and when one or more bioactive compounds are available, ligand-based virtual prescreening is utilized. This prescreening method is carried out by similarity search. The basic principle behind similarity searching is to screen databases for similar compounds with the backbone of the lead molecule. (B) In many situations, 2D similarity searches of databases are performed using chemical information from the first generation hits. (C) One alternative approach employs a ligand-based pharmacophore strategy that is often partnered with structure-based docking that uses a more stringent scoring matrix to determine the relative score made by matching two characters in a sequence alignment. This enhances the enrichment of initial hits and identifies the best compounds for computational evaluation, which are the second generation hits. (D) In the second phase, the molecular interactions between the target and the hits often identify ligand-based sites for optimizing these metrics for a unique molecular chemotype. (E) Computer algorithms, compounds or fragments of compounds from a database are positioned into a selected region of the structure (docking). These compounds are scored and ranked based on their steric and electrostatic interactions with the target site. (F) Structure determination of the target in complex with a promising lead from the first cycle reveals sites on the compound that can be optimized to increase potency.
Lately, pharmacophore approaches have become a quite important tool in drug discovery due to the absence of 3D structures of potential targets. Methods such as Pharmacophore Modeling and Quantitative Structure-Activity Relationship (QSAR) can give useful insights into the nature of target-ligand interactions, which in consequence result in predictive models that can be suitable for lead discovery and optimization.15
Ligand-based designing approaches rely on the knowledge of the structure of active ligands that interact with the target of interest to predict new chemical entities with similar behavior. The argument to justify the success of a new drug design is based on the features of pre-existing ligands, a concept known as molecular similarity; compounds with high structural similarity are more likely to have similar activity profiles.12 This methodology is considered an indirect approach to drug discovery, and it is usually used when the 3D structure of the target is unknown or cannot be predicted.
There are several ways to use a known active molecule or a set of them as a key pattern to screen a small molecule library. The first and simplest approach is the use of molecular descriptors or features.13 Physicochemical properties, such as molecular weight, volume, geometry, surface areas, atom types, dipole moment, polarizability, molar refractivity, octanol-water partition coefficient (log P), planar structures, electronegativity, or solvation properties•obtained from experimental measurements or theoretical models•are used as descriptors to compare the reference molecule or set of molecules with a large library of compounds at a very low cost. This task can be done efficiently using a symbolic representation of the molecule. These descriptors are encoded as bit strings indicating the presence or absence of the predefined properties, or can be further classified according to the different types of molecular representation: constitutional descriptors, count descriptors, list of structural fragments, fingerprints, graph invariants, quantum-chemical descriptors, and surface and volume descriptors.16
Another ligand-based approach, which is more precise than molecular descriptors, is the use of a pharmacophore modeled with ligands (ligand-based pharmacophore modeling). Given the set of known active compounds as a reference, the next step to the use of molecular descriptors is the addition of the 2D or 3D structure of these molecules to build what is called a pharmacophore model.
According to the International Union of Pure and Applied Chemistry (IUPAC), a pharmacophore is defined as “the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target, and to trigger (or block) its biological response.”17 Therefore, structurally overlapping in space features, such as steric interactions, positively and negatively charged groups, hydrogen bond donors and acceptors, or hydrophobic regions and aromatic rings•a pattern (not a molecule) representing the most probable characteristics and their geometrical constraints is used as a consensus model for the screening of small molecule library. Those new compounds show a high degree of complementarity with the pharmacophore, and are likely to be active against the protein target of interest; thus, they require a detailed and more fine study. This approach has been used extensively in de novo design, virtual screening, lead optimization and multitarget drug design, becoming a key computational strategy for facilitating drug discovery in the absence of a macromolecular target structure.18
QSAR method consists of finding a simple equation that can be used to calculate some property from the molecular structure of a compound. In QSAR modeling, the predictors include physicochemical properties or theoretical molecular descriptors of chemicals. As a result, a simple mathematical relationship is established. Applications of QSAR can be extended to any molecular design purpose, including prediction of different kinds of biological activities, lead compound optimization and prediction of new structural leads in drug discovery. The process of building a QSAR model is similar, apart from what type of property is being predicted. It consists of several steps, which lead to the design of new compounds with the desired activity profile.19 The first step involves the selection of a training set of compounds with their experimental activities to build a QSAR model. Ideally, each of these activities should cover the range of possible values for that activity. The next step is to compute descriptors that contain sufficient relevant information about the biological phenomenon. Despite the initial difficulty to predict in advance which descriptor variables will be valuable once descriptors have been calculated, one of them should be included in the QSAR model. A correlation coefficient gives a quantitative measure of how well each descriptor describes the activity. Thus, the descriptor with the highest correlation coefficient can be picked. Next, data analysis is needed to calculate the best mathematical expression linking together the descriptors and biological activities, in which information relating the essential features of the chemical and biological data structure is obtained. In the final step, validation and predictions for non-tested compounds will take place. An experimental validation of the model needs to be done, for example, by verifying already biologically tested compounds (test-set). If the QSAR predicts within acceptable restrictions, it may be used for a more extensive prediction of more compounds. Results should be interpreted for the proposal and design of new compounds with the desired activity outline.
3.2Structure-based methodsIn contrast to ligand-based methods, structure-based methods work directly with the 3D structure of a macromolecular target or a macromolecule-ligand complex. Both approaches rely on structural target information to determine whether a new compound is likely to bind with high affinity in the region where the interaction modifies the behavior of the protein with a following therapeutic effect. In this way, the target is used as a mold, where the interaction with any of the small molecules in the chemical library is computationally simulated. Subsequently, only those that showed a better fit in the binding site are selected.
These strategies are often used to improve the effect of known ligands using the biochemical information of the ligand-receptor interaction in order to postulate ligand refinements with small chemical modifications. If the information regarding the binding site exists, the steric complementarity of the ligand can be improved to increase the affinity for its receptor. Indeed, using the crystal structure of the complex, specific regions of the ligand that fit poorly within the active site can be targeted, and some chemical modifications to lower the energetic potential by making van der Waals interactions more negative may be postulated, improving the complementarity with the receptor. In a similar fashion, functional groups on the ligand can be changed in order to augment electrostatic complementarity with the receptor.18
3.3DockingIf the structure of the target has been solved at high resolution with X-ray or NMR and the molecular model of the binding site is precise enough, the best possible starting point in a structure-based drug design is the application of docking algorithms. Molecular docking is a molecular simulation technique widely used to research the interaction between the ligand and target. The docking process is the virtual simulation of the energetic interaction between the ligand and the target, including the prediction of the best ligand conformation and orientation within the binding site.20
Docking is a method that predicts the preferred orientation of one small molecule bound to a target, forming a stable complex. It consists of multiple steps. The process begins with the application of docking algorithms that pose small molecules within the active site of the target. Algorithms are complemented by scoring functions that are designed to predict the biological activity through the evaluation of interactions between compounds and potential targets. Early scoring functions evaluate compound fits based on calculations of approximate shape and electrostatic complementarities. Pre-selected conformers are often further assessed using more complex scoring schemes with a more detailed treatment of electrostatic and van der Waals interactions, and the inclusion of at least some solvation or entropic effects.21
Thus, docking programs have mainly three purposes. First, docking programs serve to identify potential ligands from a library of chemical compounds. Second, they can predict the binding mode of potential ligands or known ligands. Finally, using the predicted binding pose, these programs calculate putative binding affinities used as a score to identify those compounds which are more likely to bind the drug target.
Docking programs have shown to be successful in screening large chemical libraries, reducing them into a more manageable subset that is enriched for binders. In cases of true interactions, the predicted ligand pose often correlates well with experimentally solved protein-ligand complexes. While structure-based methods have led to the identification of novel drugs, binding pose prediction is considered one of its strengths.22 Since molecular docking plays a central role in predicting protein-ligand interactions it has been extensively used for drug hit discovery and lead optimization.19,20,23•34
3.4Structure-based pharmacophore modelingThe pharmacophore definition mentioned above is still valid to be applied when the information available to design a drug is the structure of the target. The protocol of structure-based pharmacophore modeling involves an analysis of the complementary chemical features of the active site and their spatial relationships, and a subsequent pharmacophore model assembled with selected features. In this case, a pharmacophore can be defined on the analysis of the target binding site (macromolecule (without ligand)-based) or based on a macromolecule-ligand-complex. The macromolecule-ligand-complex-based approach is convenient when a ligand is located at the ligand-binding site of a macromolecular target and the key interaction points between ligands and macromolecule need to be determined. Again, with the spatial arrangement of properties such as hydrogen bond acceptors and donors, basic or acid groups, partial charges, or aromatic and aliphatic hydrophobic regions in the active site, a virtual 3D mold can be defined with a much lower computational complexity than the target described at an all-atom level. This fact brings up the possibility to perform a virtual screening with a pharmacophore, doing a search over large libraries of compounds feasible at a reasonable cost and time.
Structure-based pharmacophore modeling has been extensively used for drug hit discovery and applied in the identification of novel ligands using a database searching approach.35 Twenty-seven homology models for 19 transporters and 38 predictive pharmacophore models from 15 drug transporters have been generated and published to date, yet only a few models (i.e., hPEPT1, P-gp, DAT, BCRP, and MRP1).36
4Integrated methodsRecently, there has been a trend towards integrating both structure- and ligand-based methods, which use information on the structure of the protein or the biological and physicochemical properties of bound ligands, respectively. The aim is to enhance the reliability of computer-aided drug design approaches by combining relevant information from the ligand and the protein. At the simplest, building a 3D pharmacophore to find potential ligands and performing further docking studies on the target constitutes a combined approach. These integrated approaches fall into two classes: interaction-based and docking similarity-based methods. Interaction-based methods focus on identifying the key interactions between the protein and ligand using available physicochemical data. These interactions are then used to screen small molecule libraries for compounds capable of producing such an interaction profile. In contrast, docking similarity-based methods merge structure-based docking methods with ligand similarity methods. With these combinations, virtual screening becomes very efficient and allows exploring libraries of up to 106 small molecules.37•41
5Virtual screeningBefore having the computational resources and techniques to perform a computational or rational drug design, researchers had to repeat an exasperating number of trial-and-error procedures against the targeted protein in their laboratories to test some hundreds of compounds available in chemical libraries. Many times, this challenge was only affordable for big pharmaceutical companies and institutions, being a matter of luck, expertise or intuition for the rest of laboratories. The number of possible chemical compounds in an exhaustive search should be around 106. Moreover, this amount of small molecules can only be tested and filtered through in silico or virtual screening (VS) with the use of powerful computers. The process of virtual screening consists of selecting compounds from large databases by using computational tools rather than physically screening them. Through this process, active compounds that could modulate a particular biomolecular pathway can be rapidly identified. The relevance of this technique is that the cost/benefit ratio justifies the presence of this approach in almost any drug design and development project, even though the number of different algorithms and strategies for successful virtual screening is a matter of constant debate and depends on the nature of the project.
Virtual screening offers many advantages over physical screening. It is significantly less resource-intensive and faster. In addition, even compounds that are not yet available can be first evaluated by virtual screening, and if they are found promising, they can be bought or synthesized. Thus, millions of compounds can be analyzed by virtual screening. However, it is important to keep in mind that virtual screening is still a relatively coarse filter, which particularly considers structure-based screening because the prediction of binding affinities remains one of the holy grails of computational chemistry.42 Nevertheless, several successful examples have been published and recently reviewed.43
6Successful applications of computational drug discovery and designRapid developments in CADDD technologies and methodologies have provided an environment to expedite the drug discovery process by enabling huge libraries of compounds to be screened and synthesized in short time and at a low cost. In the last years, numerous projects in the search for new or optimized drugs have successfully applied these approaches. To illustrate the relevance of the applications, some of them regarding in silico target prediction, hits identification or leads optimization are reviewed.
6.1In silico target predictionIn the Spring of 2003, a severe acute respiratory syndrome (SARS) outbreak occurred in China. Chen et al. identified by docking-based VS that an old drug, cinanserin, a serotonin antagonist, was a potential inhibitor of the 3C-like (3CL) protease of SARS,44 which is important in SARS coronavirus replicase polyprotein processing. The following experimental tests showed that cinanserin could indeed inhibit 3CL protease at nontoxic drug concentrations (IC50=5mM), and has the potential to kill the SARS virus. The authors concluded that because it was an old cheap drug and had an established safety record, cinanserin could be used as an emergency treatment or for stockpiling for future SARS pandemics.
Fan et al. conducted another case study of in silico target prediction in 2012. These authors established a system biology approach by combining a human reassembled signaling network with microarray gene expression data to study drug-target interactions and provide unique insights into the off-target adverse effects for torcetrapib. The results suggested that platelet-derived growth factor receptor (PDGFR), interleukin-2 (IL-2), hepatocyte growth factor receptor (HGFR) and epidermal growth factor receptor (ErbB1) tyrosine kinase were highly relevant to unfavorable effects.45 Furthermore, the obtained potential off-targets of torcetrapib were identified by employing the reverse docking strategy.
Another case study included fibroblast growth factor receptors (FGFRs), which consist of an extracellular ligand domain composed of a single transmembrane helix domain, three immunoglobulin-like domains and an intracellular domain with tyrosine kinase activity that are targets for the treatment of various human cancers. Chen et al. used the reverse pharmacophore mapping approach to identify target candidates for an active compound that they previously synthesized and showed significant in vitro antiproliferative effects. In silico target prediction revealed that tyrosine kinases might be the potential targets of the representative compound. After following structural optimization, the structure-activity relationship (SAR) analysis aided by molecular docking simulation in the ATP-binding site demonstrated that acenaphtho[1,2-b]pyrrole carboxylic acid esters are potent inhibitors of FGFR1 with IC50 values ranging from 19 to 77nM exhibited favorable growth inhibition property against FGFR-expressing cancer cell lines.46
6.2Cases of lead discovery and optimizationA lead compound has the desired activity found in a screening process, but its activity needs to be confirmed upon retesting. For lead discovery, docking (a process that involves the prediction of ligand conformation and orientation within a targeted binding site) is one of the most widely employed techniques, and it is usually embedded in the workflow of different in silico approaches. The identification of small molecules and the process of transforming these into high-content lead series are key activities in modern drug discovery.
Three compounds of the most representative examples of lead discovery and optimization are zanamivir, dorzolamide, and captopril.
Zanamivir (Relenza¨r), Gilead Sciences) is a neuraminidase inhibitor used in the treatment and prophylaxis of influenza caused by influenza A and B viruses. When the structure of the influenza neuraminidase protein was determined by X-ray crystallography, the topology of the active site was elucidated allowing for the first time the design of an inhibitor preventing the virus escaping its host cell to infect others. This achievement was accomplished using a structure-based drug design approach. Various sialic acid analogs were developed, aided by computer-assisted modeling of the active site.47
On the other hand, dorzolamide (Trusopt¨r), Merck), a carbonic anhydrase inhibitor and an anti-glaucoma agent that decreases the production of aqueous humor, was the first drug in human therapy that resulted from structure-based drug design and ab initio calculations. To achieve the design of dorzolamide successfully, the inclusion of two concepts in the project was crucial: the prototype compound generates two enantiomers, and the active-site cavity is amphiphilic.
Finally, the design of the antihypertensive drug captopril (Capoten¨r), Bristol-Myers-Squibb)•an angiotensin-converting enzyme (ACE) inhibitor used for the treatment of some types of congestive heart failure and hypertension•is an example of the early endeavors and successes of structure-based and ligand-based drug design.48 The information necessary for the design of captopril included the knowledge that the enzymatic mechanism of ACE was similar to that of carboxypeptidase A•with the difference that ACE cleaves off a dipeptide, while the carboxypeptidase A cleaves a single amino acid residue from the carboxyl end of the protein.49 Structure-activity relationship (SAR) studies were critical in guiding the synthesis of captopril 4 (IC50=23nM).50,51
7Successful applications in cancer drug discoveryThe development of new anticancer drugs proves to be a very elaborate, costly and time-consuming process. CADDD is becoming increasingly important, given the advantage that much less investment in technology, resources, and time are required. Due to the dramatic increase of information available on genomics, small molecules, and protein structures, computational tools are now being integrated at almost every stage of the drug discovery and development. Given the 3D structure of a target molecule, chemical compounds may have a potentially higher affinity for their target when are designed rationally with the aid of computational methods. In recent years, several cases of successful applications of structure-based drug design have been reported.
An interesting example of structure-based pharmacophore modeling is the identification of p53 upregulated modulator of apoptosis (PUMA) inhibitors.52 PUMA is a pro-apoptotic protein, member of the Bcl-2 protein family. Its expression is regulated by the tumor suppressor p53.53,54 PUMA ablation or inhibition leads to apoptosis deficiency underlying increased risks for cancer development and therapeutic resistance. This cancer-treatment target is central in mitochondria-mediated cell death by interacting with all known antiapoptotic Bcl-2 family members.54 Based on the binding of BH3-only proteins with Bcl-2-like proteins, some approaches have been used to identify small molecules that can modulate these interactions and therefore, inhibit apoptosis. Most of the efforts have focused on the development of Bcl-2 family inhibitors that mimic the actions of the pro-apoptotic BH3 domains. Such compounds have been identified through computational modeling, structure-based design, and high-throughput screening of natural product and synthetic libraries.55
On the other hand, Liu et al. reported a combinatorial computational strategy for discovering potential inhibitors against insulin-like growth factor-1 receptor (IGF-1R), which is associated with several cancers, including breast, prostate, and lung cancer. IGF-1R belongs to the tyrosine kinase family and plays a pivotal role in the signaling pathway involving cell growth, proliferation, and apoptosis. The initial hit obtained from hierarchical VS was subsequently used as the query scaffold for the substructure search to build a focused library. The library was then screened against IGF-1R with an in-house pharmacophore-constrained docking protocol. Eventually, 15 out of 39 compounds exhibited inhibitory activity in enzymatic assessment. Strikingly, the two most potent inhibitors demonstrated an excellent inhibitory potency (IC50=57 and 61nM, respectively), and also presented significant selectivity over the insulin receptor (IR), which is highly homologous to IGF-1R.56 The authors concluded that the promising selective IGF-1R inhibitors•aside from being potential antitumor agents•could be investigated as molecular probes to differentiate the biological functions of IGF-1R and IR.
Another successful example of small molecules designed using a ligand-based approach is the case of tubulin inhibitors.57 Tubulin polymerization, an essential component of cell cycle progression and cell division represents an important target for anticancer therapy. Several antimitotic agents (paclitaxel, colchicine, and the vinca alkaloids) have been discovered and are clinically used, but they often show significant toxicity, low bioavailability, rapidly acquired resistance and the resulting overexpression of drug-resistant pumps that eject these antimitotic inhibitors from the cell. However, due to these unfavorable properties, researchers have devoted substantial effort to discover new agents with more tolerable and efficient properties, particularly as it is believed that antimitotic agents could work to diminish blood supply to cancerous tumors.
Liou et al. based their model generation on a set of 21 indole-derivatives synthesized originally for potential tubulin inhibition and used structure-activity relationship (SAR) analysis to drive it. These compounds were chosen such that their inhibitory IC50 values spanned over three orders of magnitude, from 1.2nM to 6α/4M. Based on the chemical similarities of these compounds, the authors selected four common pharmacophoric features•including a hydrogen bond donor, a hydrogen bond acceptor, a hydrophobic group, and a hydrophobic aromatic group•for the construction of a chemical library. Subsequently, 142 compounds were biologically tested using a human oral squamous carcinoma cell line (KB). Among these 142 biologically tested compounds, four were found to inhibit the KB cell line with IC50 values of 187nM, 2.0α/4M, 3.0α/4M, and 5.7α/4M, respectively. The most potent compound of these four active molecules was also found to inhibit the proliferation of other cancer cell lines like MCF-7 (breast cancer), NCI-H460 (human non-small-cell lung cancer), and SF-268 (human central nervous system cancer), with IC50 values of 236nM, 285nM, and 319nM, respectively.58
Another example of small molecules designed using a computational approach is the case of an I-Kappa-B Kinase β (IKK-β) inhibitor.59 IKK-β, which is a key player in the NF-κB signaling pathway, represents yet another potential target for the treatment of cancer in addition to inflammation. In 2011, Noha et al. decided to use ligand-based pharmacophore modeling to identify new compounds with affinity to IKK-β. The ligand-based pharmacophore model for this study was based on a set of five compounds with high activity (IC50 values of 100nM or less) and at least a several-fold difference in selectivity for IKK-β over NFκB in an attempt to develop an IKK-β inhibitor-specific pharmacophore model. The model was further refined using a dataset extracted from the literature of 44 biologically inactive compounds, 128 active compounds, and 12,775 several random decoy compounds. The top ten high-scoring compounds were tested in vitro. Among these, the most potent inhibitor (compound NSC-719177) could inhibit IKK-β with an IC50 value of approximately 6.95α/4M.59 Cell-based analyses were also conducted to test the ability of compound NSC-719177 to inhibit NFκB activation in HEK293 cells stably transfected and carrying a luciferase reporter gene activated by a promoter composed of multiple copies of the NF-κB response element. Compound NSC-719177 was found to have a cell-based assay IC50 value of approximately 5.85α/4M and exhibited dose-dependent activity in inhibiting TNF-α-induced luciferase activity. Therefore, Noha et al. were able to demonstrate the successful application of ligand-based approaches to the identification of low micromolar inhibitors against IKK-β.59
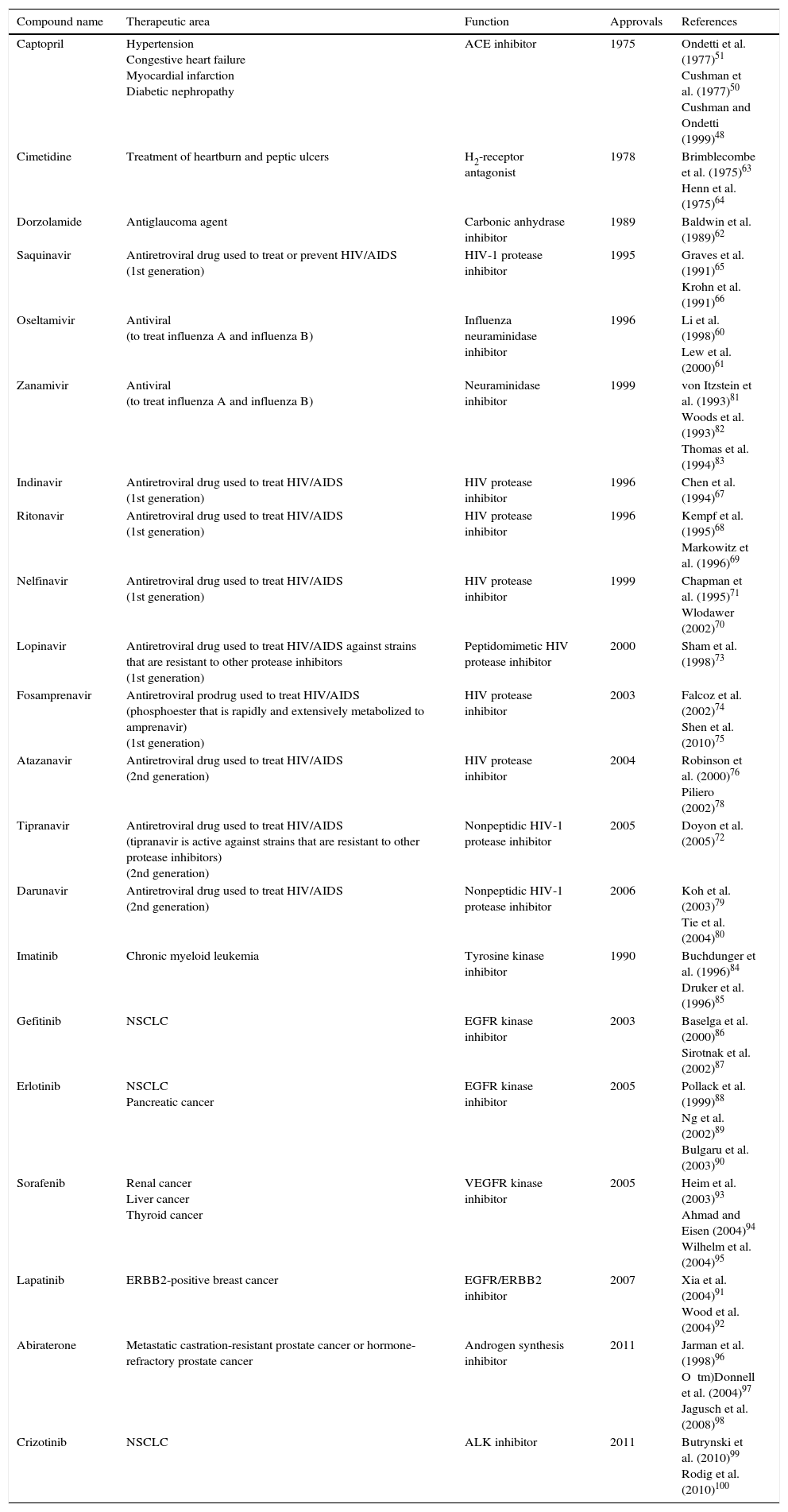
In an attempt to articulate how the iterative process of structure-based design can lead to the development of drugs with pharmacological potential, more examples from the literature are listed in Table 1.60•100
Selected inhibitors developed with computational chemistry and rational drug design strategies.
| Compound name | Therapeutic area | Function | Approvals | References |
|---|---|---|---|---|
| Captopril | Hypertension Congestive heart failure Myocardial infarction Diabetic nephropathy | ACE inhibitor | 1975 | Ondetti et al. (1977)51 Cushman et al. (1977)50 Cushman and Ondetti (1999)48 |
| Cimetidine | Treatment of heartburn and peptic ulcers | H2-receptor antagonist | 1978 | Brimblecombe et al. (1975)63 Henn et al. (1975)64 |
| Dorzolamide | Antiglaucoma agent | Carbonic anhydrase inhibitor | 1989 | Baldwin et al. (1989)62 |
| Saquinavir | Antiretroviral drug used to treat or prevent HIV/AIDS (1st generation) | HIV-1 protease inhibitor | 1995 | Graves et al. (1991)65 Krohn et al. (1991)66 |
| Oseltamivir | Antiviral (to treat influenza A and influenza B) | Influenza neuraminidase inhibitor | 1996 | Li et al. (1998)60 Lew et al. (2000)61 |
| Zanamivir | Antiviral (to treat influenza A and influenza B) | Neuraminidase inhibitor | 1999 | von Itzstein et al. (1993)81 Woods et al. (1993)82 Thomas et al. (1994)83 |
| Indinavir | Antiretroviral drug used to treat HIV/AIDS (1st generation) | HIV protease inhibitor | 1996 | Chen et al. (1994)67 |
| Ritonavir | Antiretroviral drug used to treat HIV/AIDS (1st generation) | HIV protease inhibitor | 1996 | Kempf et al. (1995)68 Markowitz et al. (1996)69 |
| Nelfinavir | Antiretroviral drug used to treat HIV/AIDS (1st generation) | HIV protease inhibitor | 1999 | Chapman et al. (1995)71 Wlodawer (2002)70 |
| Lopinavir | Antiretroviral drug used to treat HIV/AIDS against strains that are resistant to other protease inhibitors (1st generation) | Peptidomimetic HIV protease inhibitor | 2000 | Sham et al. (1998)73 |
| Fosamprenavir | Antiretroviral prodrug used to treat HIV/AIDS (phosphoester that is rapidly and extensively metabolized to amprenavir) (1st generation) | HIV protease inhibitor | 2003 | Falcoz et al. (2002)74 Shen et al. (2010)75 |
| Atazanavir | Antiretroviral drug used to treat HIV/AIDS (2nd generation) | HIV protease inhibitor | 2004 | Robinson et al. (2000)76 Piliero (2002)78 |
| Tipranavir | Antiretroviral drug used to treat HIV/AIDS (tipranavir is active against strains that are resistant to other protease inhibitors) (2nd generation) | Nonpeptidic HIV-1 protease inhibitor | 2005 | Doyon et al. (2005)72 |
| Darunavir | Antiretroviral drug used to treat HIV/AIDS (2nd generation) | Nonpeptidic HIV-1 protease inhibitor | 2006 | Koh et al. (2003)79 Tie et al. (2004)80 |
| Imatinib | Chronic myeloid leukemia | Tyrosine kinase inhibitor | 1990 | Buchdunger et al. (1996)84 Druker et al. (1996)85 |
| Gefitinib | NSCLC | EGFR kinase inhibitor | 2003 | Baselga et al. (2000)86 Sirotnak et al. (2002)87 |
| Erlotinib | NSCLC Pancreatic cancer | EGFR kinase inhibitor | 2005 | Pollack et al. (1999)88 Ng et al. (2002)89 Bulgaru et al. (2003)90 |
| Sorafenib | Renal cancer Liver cancer Thyroid cancer | VEGFR kinase inhibitor | 2005 | Heim et al. (2003)93 Ahmad and Eisen (2004)94 Wilhelm et al. (2004)95 |
| Lapatinib | ERBB2-positive breast cancer | EGFR/ERBB2 inhibitor | 2007 | Xia et al. (2004)91 Wood et al. (2004)92 |
| Abiraterone | Metastatic castration-resistant prostate cancer or hormone-refractory prostate cancer | Androgen synthesis inhibitor | 2011 | Jarman et al. (1998)96 O tm)Donnell et al. (2004)97 Jagusch et al. (2008)98 |
| Crizotinib | NSCLC | ALK inhibitor | 2011 | Butrynski et al. (2010)99 Rodig et al. (2010)100 |
ACE, angiotensin-converting enzyme; HIV, human immunodeficiency virus; AIDS, acquired immunodeficiency syndrome; EGFR, epidermal growth factor receptor; NSCLC, non-small cell lung cancer; VEGFR, vascular epidermal growth factor receptor; ERBB2, erb-b2 receptor tyrosine kinase 2 (also known as NEU, NGL, HER2, TKR1, CD340, HER-2, MLN 19, HER-2/neu); ALK, anaplastic lymphoma kinase.
With the evolution of significantly more sophisticated molecular modeling tools and the use of high-throughput X-ray crystallography for a target alone or in complex with small molecules, rational drug design techniques have become an indispensable instrument for the development of target-based therapies.
Considering the relevance of the examples cited above, and the importance of these approaches, our research group is interested in finding some molecules capable of inhibiting the transcription factor called Yin-Yang 1 (YY1). This factor has been observed to be overexpressed in cancer patients, and its activation is significantly involved in chemotherapy resistance mechanisms. In pediatric patients with acute lymphoblastic leukemia (ALL) it was reported that YY1 is overexpressed.101 The in vitro inhibition in ALL cell lines showed that YY1 has an important implication in chemoresistance in these cells. Based on these findings, our research group was particularly interested in identifying some inhibitors of the activity of this transcription factor employing CADDD tools. The elucidation of the structure of the YY1 and site-binding domain provides a new framework to understand the functions of this transcription factor and leads to the development of rational drug design for the treatment of ALL. An overview of YY1 structure and activity, its actions in lymphoblastic leukemia and chemoresistance, and how structural information and high-throughput screening have been or can be used for drug discovery will be provided in the future.
The process of novel drug discovery and development is recognized to be very expensive and time-consuming. However, thanks to recent advances in the development of physical and chemical models to simulate biomolecular processes, together with the production of increasingly powerful computational resources, discovering and designing new drugs as anticancer drugs is an affordable task for many research institutions and laboratories today. With the required computational hardware and software, and the expertise in biochemistry, biophysics, and biology, many projects that previously demanded a significant investment in time and money can be done today by a small group of researchers in their workstations. Moreover, challenging projects not even conceivable two decades ago can be today tackled with the access to a supercomputer. By using CADDD approaches, researchers are not only accelerating their steps and projects. Implementing molecular simulations in biomolecular research projects has increased our knowledge in fields such as structural and chemical biology at the point where these tools are considered as other useful facilities in the laboratory. The optimization of these techniques and methods occurs this way naturally in its theoretical feedback signaling system. Computational models generate useful predictions to be checked with experimental results, and biologists and physicians demand approaches that are more accurate to computational scientists.
Conflict of interestThe authors declare no conflicts of interest of any nature.