



Wilson's disease (WD), resulting from homozygote and compound heterozygote mutations in ATB7B, is an autosomal recessive disease. WD associated acute liver failure (ALF) is fatal, and a revised Wilson's disease prognostic index (RWPI) >11 is a reliable indication of liver transplantation (LT) or artificial liver support (ALS). We described a WD patient who initially presented with ALF and severe hemolytic anemia. A single heterozygote c.2333G>T mutation (p. Arg778Leu, R778L) in ATP7B was screened by whole exome sequencing and validated by Sanger sequencing. Rapid diagnostic criteria (ALP/TBIL <4 and AST/ALT >2.2) are suitable for early diagnosis. Although the RWPI amounted to 15, the patient recovered after intermittent plasma transfusion and subsequent chelating therapy without LT or ALS. In conclusion, WD patients with a single R778L heterozygote mutation can present with ALF as the initial clinical manifestation, and intermittent plasma transfusion combined with chelating therapy may alleviate fulminant WD without LT or ALS.
Wilson's disease (WD) is an autosomal recessive disease, resulting from ATP7B gene mutations and copper accumulation in the liver and extrahepatic tissues. WD's manifestations may range from an asymptomatic state to life-threatening acute liver failure (ALF), with or without neurological presentations. The prevalence of WD is estimated at 1:30,000 to 5.87:100,000, with a heterozygote carrier frequency of approximately 1% [1]. It was reported that c.2333G>T (p.Arg778Leu, R778L) homozygote and compound heterozygote mutations of ATP7B are the most common alterations in the Chinese population [2]. However, the associations of genotypes with phenotypes in WD patients with single R778L heterozygote mutations remain undefined.
Moreover, ALF mortality due to WD approaches 100%, and liver transplantation (LT) is the only effective treatment for patients with WD who present with ALF [1,3,4]. Revised Wilson's disease prognostic index (RWPI)>11 [5,6] is a reliable indication for LT. Meanwhile, combining plasma exchange and chelating therapy has been reported to rescue ALF in Wilson's disease without liver transplantation [7]. Several reports showed that advances of artificial liver support (ALS) may improve the outcome of ALF [8]. However, LT is limited by donor shortage, and ALS remains unavailable in some areas because of costly equipment and the lack of plasma.
We herein described a case of WD harboring a single R778L heterozygote mutation, and initially presenting with acute liver failure and severe hemolytic anemia. The patient recovered after intermittent plasma transfusion and subsequent chelating therapy without LT or ALS.
2Case presentationThe patient was a 13-year-old Chinese girl admitted to the Liver Department of the Third People's Hospital of Changzhou on 17 January 2017. She presented with a 10-day history of fatigue, nausea, and loss of appetite. She had no history of drug or alcohol abuse. Family history was negative for metabolic and inherited liver diseases. Her parents had no consanguineous marriage. On physical examination, the girl had pronounced jaundice on the skin and sclera, with ecchymosis at the injection sites. Palpable liver and splenomegaly were found by abdominal ultrasound. Cornea Kayser-Fleischer rings were found using slit lamp examination. The clinical data of the patient were obtained during one year of follow-up. The current study was approved by the ethics committee of the Third People's Hospital of Changzhou and performed according to the Declaration of Helsinki in 1975. Informed consent was obtained from the girl and her parents.
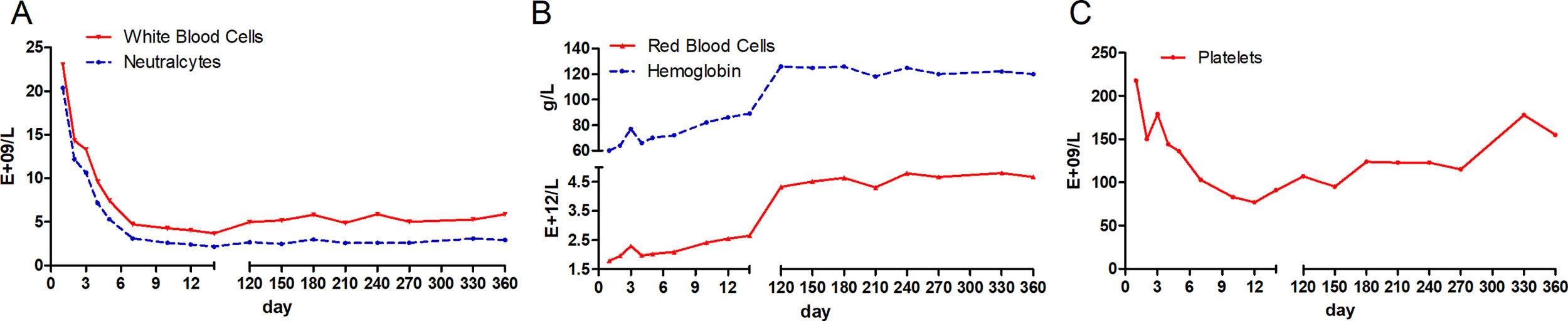
At the time of admission, laboratory assessment revealed abnormal liver function tests, Coomb's negative hemolytic anemia and coagulopathy. Bone marrow morphology showed hyperplastic anemia. She had aspartate amino-transaminase (AST) levels nearly 2 times the upper limit of the normal range (104U/L), whereas alanine aminotransferase (ALT) amounts were normal (27U/L). Total bilirubin (TBIL) was more than 35 times the upper limit of the normal range (613.1μmol/L), with the majority being unconjugated bilirubin (416.1μmol/L). Liver failure was diagnosed according to overtly altered TBIL (613.1μmol/L), international normalized ratio (INR, 2.62) and prothrombin activity (24.27%); the RWPI score amounted to 15 [6]. Moreover, the hemogram test showed high levels of white blood cells (23.3E+9/L) and severe anemia (hemoglobin, 60g/L). Serological markers for HBV, HAV, HCV, HDV and HEV were all negative except for anti-HBs.
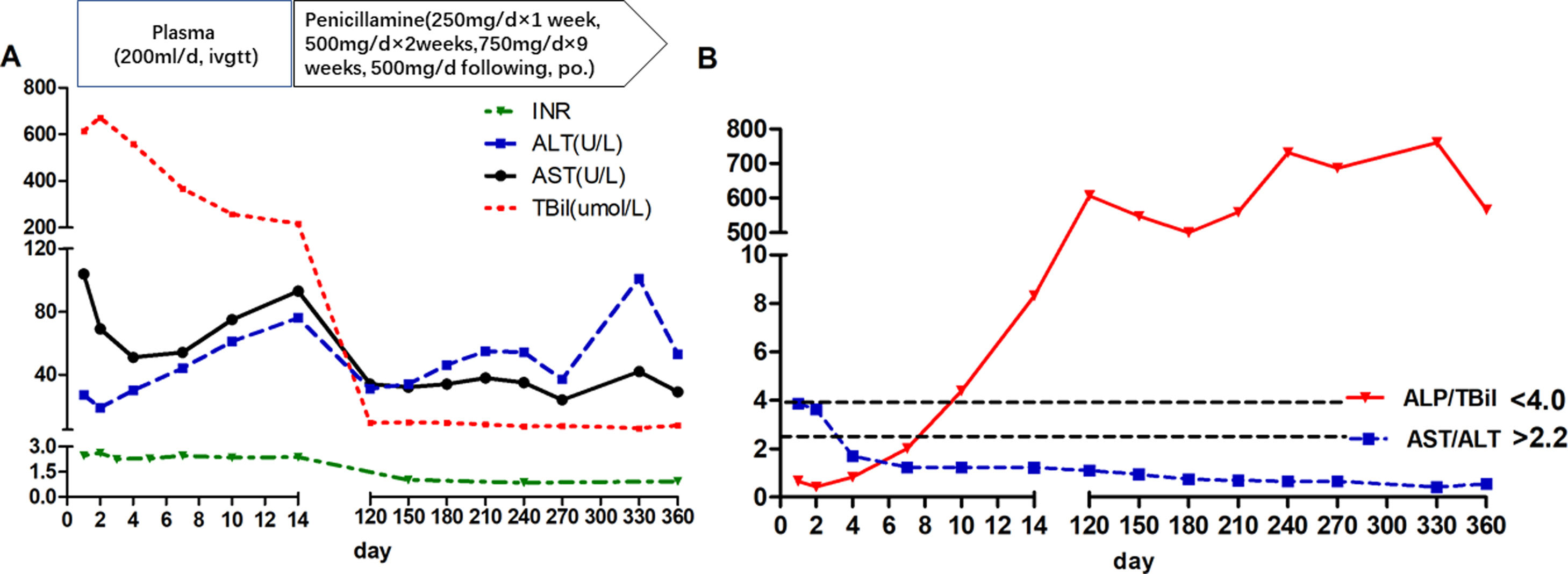
During the first 14 days after onset, fresh plasma was administered intravenously every day at a dose of 200ml, and erythrocytes were transfused every other day at a dose of 2U. TBIL was overtly decreased at the 14th day after admission (Fig. 1A). In addition, white blood cell count was reduced, while anemia was ameliorated (Fig. 2). Ceruloplasmin was low (0.04g/L) and WD was considered; then, the patient was transferred to the First Hospital of Anhui Traditional Chinese Medicine University. Penicillamine was used as a copper-chelating drug and administrated on the 15th day after admission (250mg/d for the 1st week, then 500mg/d for 2 weeks, 750mg/d for 9 weeks, and 500mg/d for subsequent months, continued till now).
 and respective ratios (B).")
, red blood cells (B) and platelet (C).")
During the one-year follow-up, TBIL was maintained below 10μmol/L, whereas alkaline phosphatase (ALP) levels increased to 258U/L at the 4th month and lasted for several months. On December 19, 2017 (330th day after onset), further tests revealed a slight elevation of ALT (101U/L), which declined a month later.
The ALP (U/L) to TBIL (mg/dl) ratio was lower than 4, and that of AST (U/L) to ALT (U/L) was higher than 2.2 at admission; meanwhile, the ALP/TBIL ratio increased to above 4 at day 10 after admission, and AST/ALT declined to below 2.2 at day 4. After one year of follow-up, ALP was maintained at a high level (above 258U/L) upon recovery, while the ALP/TBIL ratio was increased to 607 and lasted for several months. The AST/ALT ratio remained <2.2 until the end of follow-up (Fig. 1B).
As whole exome sequencing is attractive in diagnosing patients with life-threatening liver diseases of indeterminate etiology [9], it was performed for this patient. Genome DNA was extracted with QIAamp DNA Blood Kit (Qiagen, Tokyo, Japan). The entire genomic DNA was amplified with adapter-modified fragments; then, a DNA library was established according to the manufacturer's instructions (Agilent, Santa Clara, CA, USA) and captured using Agilent's SureSelect Human All Exon V6 Kit. DNA fragments were sequenced on an Illumina HiSeq X Ten for paired-end 150bp reads. Raw reads were processed to remove adaptor sequences and trim low-quality reads (base quality <30), and aligned to the reference genome sequence (UCSC hg19) with Bowtie2 version 2.2.6 [10]. Mutations were then detected with GATK [11], Lofreq [12] and VarScan [13] from aligned BAM files. Databases, including dbSNP, OMIM and ClinVar, and annotation software, including SIFT, PolyPhen, MutationTaster, Provean and MetaSVM, were used to screen potential disease associated mutations. The ATP7B gene fragments which contained mutations screened in whole exome sequencing were amplified by PCR and sequenced after purification.
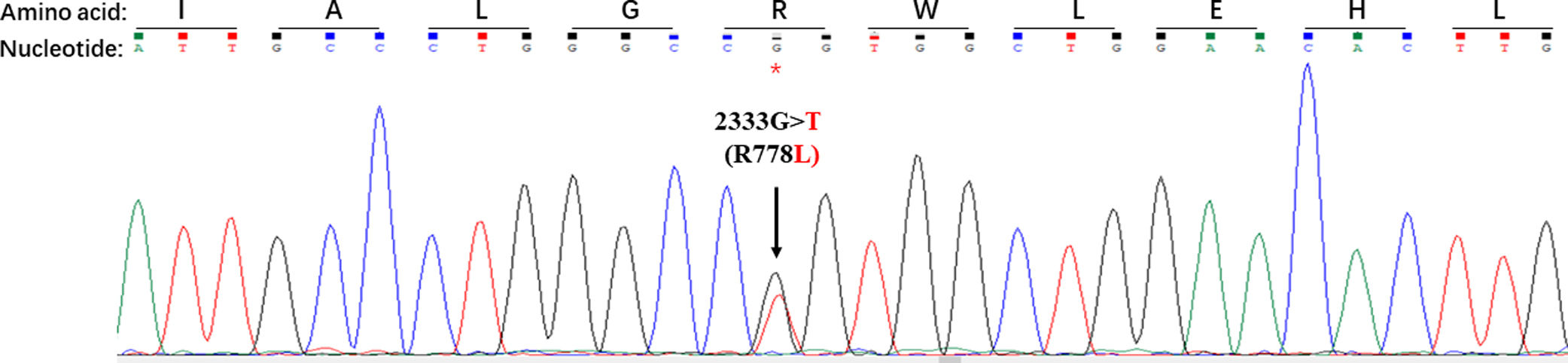
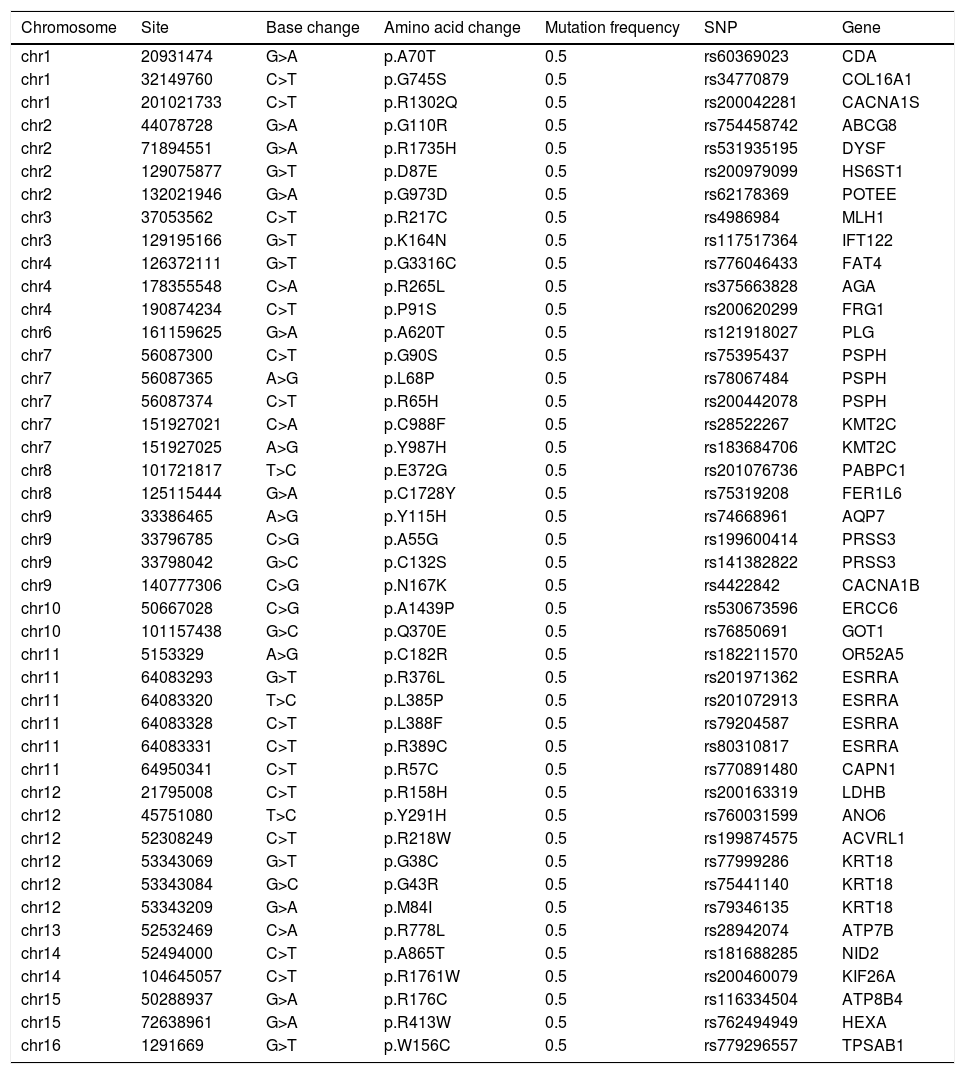
As shown in Table 1, a total of 44 disease causing variations were screened. A single R778L heterozygote mutation in the ATP7B gene was identified by whole exome sequencing and validated by Sanger sequencing (Fig. 3).
Disease causing variations (n=44) screened by whole exome sequencinga.
| Chromosome | Site | Base change | Amino acid change | Mutation frequency | SNP | Gene |
|---|---|---|---|---|---|---|
| chr1 | 20931474 | G>A | p.A70T | 0.5 | rs60369023 | CDA |
| chr1 | 32149760 | C>T | p.G745S | 0.5 | rs34770879 | COL16A1 |
| chr1 | 201021733 | C>T | p.R1302Q | 0.5 | rs200042281 | CACNA1S |
| chr2 | 44078728 | G>A | p.G110R | 0.5 | rs754458742 | ABCG8 |
| chr2 | 71894551 | G>A | p.R1735H | 0.5 | rs531935195 | DYSF |
| chr2 | 129075877 | G>T | p.D87E | 0.5 | rs200979099 | HS6ST1 |
| chr2 | 132021946 | G>A | p.G973D | 0.5 | rs62178369 | POTEE |
| chr3 | 37053562 | C>T | p.R217C | 0.5 | rs4986984 | MLH1 |
| chr3 | 129195166 | G>T | p.K164N | 0.5 | rs117517364 | IFT122 |
| chr4 | 126372111 | G>T | p.G3316C | 0.5 | rs776046433 | FAT4 |
| chr4 | 178355548 | C>A | p.R265L | 0.5 | rs375663828 | AGA |
| chr4 | 190874234 | C>T | p.P91S | 0.5 | rs200620299 | FRG1 |
| chr6 | 161159625 | G>A | p.A620T | 0.5 | rs121918027 | PLG |
| chr7 | 56087300 | C>T | p.G90S | 0.5 | rs75395437 | PSPH |
| chr7 | 56087365 | A>G | p.L68P | 0.5 | rs78067484 | PSPH |
| chr7 | 56087374 | C>T | p.R65H | 0.5 | rs200442078 | PSPH |
| chr7 | 151927021 | C>A | p.C988F | 0.5 | rs28522267 | KMT2C |
| chr7 | 151927025 | A>G | p.Y987H | 0.5 | rs183684706 | KMT2C |
| chr8 | 101721817 | T>C | p.E372G | 0.5 | rs201076736 | PABPC1 |
| chr8 | 125115444 | G>A | p.C1728Y | 0.5 | rs75319208 | FER1L6 |
| chr9 | 33386465 | A>G | p.Y115H | 0.5 | rs74668961 | AQP7 |
| chr9 | 33796785 | C>G | p.A55G | 0.5 | rs199600414 | PRSS3 |
| chr9 | 33798042 | G>C | p.C132S | 0.5 | rs141382822 | PRSS3 |
| chr9 | 140777306 | C>G | p.N167K | 0.5 | rs4422842 | CACNA1B |
| chr10 | 50667028 | C>G | p.A1439P | 0.5 | rs530673596 | ERCC6 |
| chr10 | 101157438 | G>C | p.Q370E | 0.5 | rs76850691 | GOT1 |
| chr11 | 5153329 | A>G | p.C182R | 0.5 | rs182211570 | OR52A5 |
| chr11 | 64083293 | G>T | p.R376L | 0.5 | rs201971362 | ESRRA |
| chr11 | 64083320 | T>C | p.L385P | 0.5 | rs201072913 | ESRRA |
| chr11 | 64083328 | C>T | p.L388F | 0.5 | rs79204587 | ESRRA |
| chr11 | 64083331 | C>T | p.R389C | 0.5 | rs80310817 | ESRRA |
| chr11 | 64950341 | C>T | p.R57C | 0.5 | rs770891480 | CAPN1 |
| chr12 | 21795008 | C>T | p.R158H | 0.5 | rs200163319 | LDHB |
| chr12 | 45751080 | T>C | p.Y291H | 0.5 | rs760031599 | ANO6 |
| chr12 | 52308249 | C>T | p.R218W | 0.5 | rs199874575 | ACVRL1 |
| chr12 | 53343069 | G>T | p.G38C | 0.5 | rs77999286 | KRT18 |
| chr12 | 53343084 | G>C | p.G43R | 0.5 | rs75441140 | KRT18 |
| chr12 | 53343209 | G>A | p.M84I | 0.5 | rs79346135 | KRT18 |
| chr13 | 52532469 | C>A | p.R778L | 0.5 | rs28942074 | ATP7B |
| chr14 | 52494000 | C>T | p.A865T | 0.5 | rs181688285 | NID2 |
| chr14 | 104645057 | C>T | p.R1761W | 0.5 | rs200460079 | KIF26A |
| chr15 | 50288937 | G>A | p.R176C | 0.5 | rs116334504 | ATP8B4 |
| chr15 | 72638961 | G>A | p.R413W | 0.5 | rs762494949 | HEXA |
| chr16 | 1291669 | G>T | p.W156C | 0.5 | rs779296557 | TPSAB1 |

The present study assessed a WD patient harboring a single R778L heterozygote mutation and initially presenting with ALF and severe hemolytic anemia. Interestingly, the patient recovered after intermittent plasma transfusion and subsequent chelating therapy without LT or ALS.
Approximately 5% of WD patients develop ALF, and up to 50% of pediatric patients with ALF do not show encephalopathy [14–16]. The current patient had evidence of severe liver injury (TBIL 613.1μmol/L) and coagulopathy (INR=2.62) at admission, and she met ALF criteria according to a previous report [6], in spite of the absence of encephalopathy. The RWPI score of the patient amounted to 15 (TBIL>301μmol/L; INR>2.5; AST>100U/L; albumin<33g/L; white blood count>15.4E+09/L), with >11 representing a reliable indicator of LT [6]. Of these parameters, elevated white blood cell count is considered a marker of occult infection, a stress response to liver injury or an unidentified factor that predicts liver failure severity. Considering that procalcitonin is normal and no definite infectious focus exists, there was insufficient evidence of infection or sepsis for this patient.
The Leipzig [17] and rapid diagnostic [18,19] criteria can be used for ALF patients with suspected WD. According to Leipzig criteria, this patient had a cumulative score of 6, with a score >4 indicating reliable diagnosis of WD. The rapid diagnostic criteria [17], including ALP/TBIL below 4 and AST/ALT above 2.2, have been reported to have 100% sensitivity and specificity in WD diagnosis. For this patient, ALP/TBIL and AST/ALT ratios met these criteria at admission, while ALP/TBIL increased to above 4 at day 10 after admission; meanwhile, AST/ALT fell below 2.2 at day 4 and lasted until the end of follow-up. Notably, the diagnostic criteria are suitable for early diagnosis since these ratios change during the disease course.
Disease causing homozygote mutations can support WD diagnosis with certainty, whereas heterozygote mutations with clinical symptoms are scarce [17]. To the best of our knowledge, a single R778L heterozygote mutation in the ATP7B gene in patients with ALF has not been reported. In the present study, only a single R778L heterozygote mutation in the ATP7B gene was detected by whole exome sequencing and validated by Sanger sequencing. These findings confirmed that a WD patient carrying a single R778L heterozygote mutation can present with ALF as initial clinical manifestations.
In addition, liver transplantation is considered the only life-saving option for fulminant WD patients after failure of copper-chelating therapy [4]. Remission can also be achieved by combining ALS and chelating therapy in some cases [7,8]. Considering that liver function and the general condition were overtly improved before copper-chelating therapy, we presume that plasma transfusion combined with penicillamine may alleviate fulminant WD in the transitional period when LT or ALS is unavailable. However, the role of plasma transfusion in WD treatment remains elusive and requires further investigation to support the above notion. Undoubtedly, liver transplantation and ALS are still the preferred approaches for fulminant WD.
In conclusion, a WD patient carrying a single R778L heterozygote mutation can present with ALF as initial clinical manifestations, and rapid diagnostic criteria (ALP/TBIL<4 and AST/ALT>2.2) are suitable for early diagnosis. Moreover, intermittent plasma transfusion and copper-chelating therapy may rescue fulminant WD when LT or ALS is unavailable.AbbreviationsWD Wilson's disease acute liver failure alanine aminotransferase aspartate aminotransferase alkaline phosphatase total bilirubin international normalized ratio liver transplantation artificial liver support revised Wilson disease prognostic index
Yuan Xue conceived and designed the study. Longgen Liu, Qing Gong, Juan Liu, Hongyu Shen and Hongyu Zhang collected and confirmed the data. Longgen Liu and Qing Gong analyzed the data and drafted the manuscript. Yuan Xue revised of the manuscript. All authors read and approved the final manuscript. Longgen Liu and Qing Gong contributed equally to this work.
Financial supportThis work was supported by grants from the Chinese Foundation for Hepatitis Prevention and Control-Tianging Liver Disease Research Fund Subject (TQGB20150006 and TQGB201700139) and the Science and Technology Project of Changzhou (CJ20160024).



