



Pediatric Systemic Lupus Erythematosus (pSLE) is an autoimmune disorder of children. Early disease onset raises the probability of genetic etiology and it is more severe than adult SLE.
Patients and methodsHerein an eight-year-old girl with pSLE from consanguineous parents is reported.
ResultsAlthough she was diagnosed as pSLE since the age of two years, Whole Exome Sequencing (WES) revealed a rare stop-gained C>T mutation in C1QA gene. The variant was validated and segregated in patient and the family. Furthermore, serum levels of the C1q protein were measured and found to be much lower than normal ranges.
ConclusionsThis study indicated that C1Q deficiency should be considered as a differential diagnosis of pSLE. Therefore, measurement of C1q should be recommended in all cases with pSLE.
Systemic Lupus Erythematosus (SLE) is a systemic autoimmune disease that affects many organs such as skin, kidneys and joints. About 10–20% of this autoimmunity occurs in children younger than 18 years, which refers as Pediatric SLE (pSLE). Compared to SLE in adults, the current knowledge on pSLE is scarce.1,2 pSLE usually occurs below 10–12 years of age and disease onset prior to this age has been rarely reported.3 Unfortunately, children affected by SLE have a worse course of disease and more devastating prognosis compared to adults.2 SLE complications such as nephritis, hematologic disorders, mucosal ulcers and skin rashes (butterfly rash) are more frequent in pediatric onset than adult onset of disease.2 Neuropsychiatric complications such as seizure and psychosis are reported in about 25% of patients and may be the first presenting manifestation of pSLE.4
Although the exact underlying pathogenesis of pSLE is not yet well understood, both genetic and environmental factors have been suggested to be important. Genetic defects are more likely to be seen in patients with pediatric early onset SLE compared to the adult onset form of the disease. This highlights the need for more investigations on possible genetic factors underlying pSLE that may be of value in both diagnostic and future therapeutic avenues.
It has previously been reported that SLE could be considered as a monogenic disorder in a proportion of cases, which occurs as a result of mutations in different genes.5 These genes are usually involved in different cellular pathways such as apoptosis, nucleic acid degradation and repair, B cell development, and pro-inflammatory cytokine pathways (i.e. IFN I); and they can affect normal tolerance. Autosomal recessive PRKCD and DNASE1L3 mutations have been reported as the causative gene defects in patients with pSLE.6,7 Additional genes that are involved in DNA degradation pathway such as TREX-1 and DNASE1 can cause autosomal dominant SLE with early age of onset.8,9 Other gene defects such as IFIH1 gain of function and ACP5 loss of function mutations were also reported to be the genetic causes of severe early-onset SLE.10,11 Early components of the complement system have also been reported to cause monogenic SLE-like syndrome.12 Here, we report a case of pSLE with C1Q mutation that occurred as early as 20 months of age.
MethodsSample preparationPeripheral blood sample was collected from the patient with the consent of her parents. In addition to the patient, samples from parents and healthy sibling(s) were also collected; all family members signed informed consent before enrolment in the study. This study was approved by the Ethics Committee of Tehran University of Medical Sciences. Genomic DNA was extracted from all samples by standard phenol chloroform method. Serum samples were separated and kept in −70°C for further protein analysis.
Whole Exome Sequencing (WES)TrueSeq Rapid Exome kit was used for genome library preparation, after cluster generation; sequencing was performed using Illumina HiSeq3000 platform. After sequencing, readings were aligned to the human genome version 19 using the Burrows-Wheeler Aligner (BWA). Variant annotation and functional effect prediction was performed using the SNPEFF tool. The variant list was then filtered according to the minor allele frequency (MAF) >0.01 in ExAC browser. Furthermore, an internal database was used to characterize recurrent variants. Finally, combined annotation dependent depletion (CADD) score was used for prediction of the effects of variants in order to prioritize them.13 Homozygosity mapping was performed by means of H3M2 (Homozygosity Heterogeneous Hidden Markov Model) software, which allows the detection of runs of homozygosity from whole-exome sequencing data.14
Evaluation of C1q serum levelIn order to evaluate the serum level of C1q protein, an ELISA kit was used (Abcam, UK) according to the manufacturer's protocol.
ResultsPatient historyThe case is an eight-year-old girl born from a consanguine Iranian family, who was primarily referred to Children's Medical Center hospital, a tertiary referral center affiliated to Tehran University of Medical Sciences at the age of two years. The patient started her first manifestation at the age of 20 months, by ataxic gait and imbalance. The gait disorder regressed in a period of four months, but her growth and development were completely normal before the disease onset. Furthermore, the patient experienced fever and seizure. Prolongation of fever, despite antibiotic therapy, led to some rheumatologic work ups, which revealed positive Anti-Nuclear Antibody (ANA). The patient gradually lost her walking and standing abilities, leading to walking disability at the time of her first admission. In physical examination, her extremity muscles were spastic. Later on, she developed malar rash and oral ulcers. Transverse myelitis was considered primarily as a potential reason for the patient's movement disorder; however, it was not proved since spinal magnetic resonance imaging (MRI) and cerebrospinal fluid (CSF) analysis were normal. Electromyography (EMG) and nerve conduction velocity (NCV) were also performed for the patient in order to detect myopathy or neuropathy, which were normal as well.
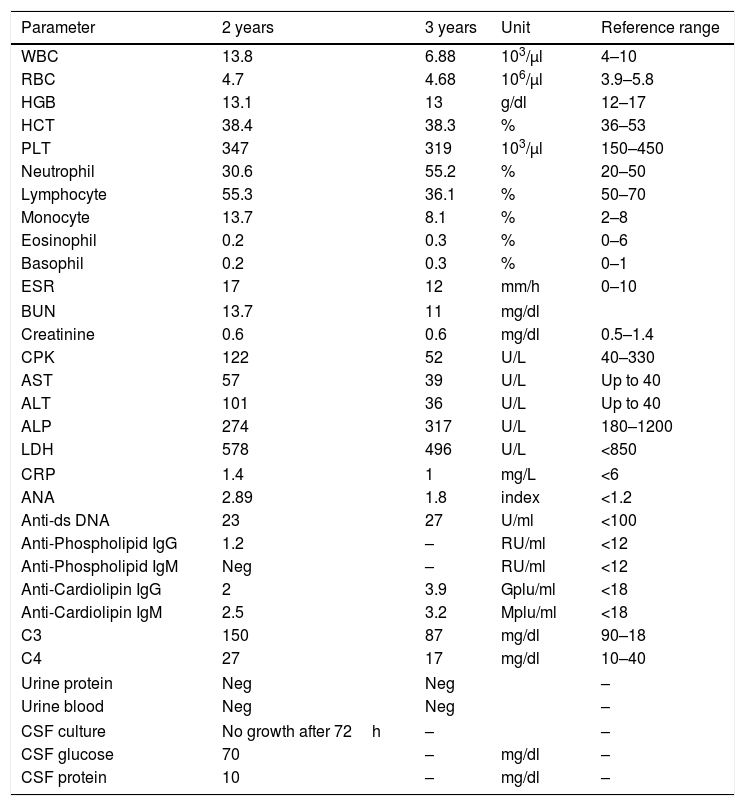
In laboratory investigations, the patient had positive titer of Anti-dsDNA once in her medical record, which was not repeated again. C3 and C4 serum levels were normal. Her lab results are summarized in Table 1. The patient was hospitalized again at the age of three and six years, because of feet spasticity, skin lesions, painful oral ulcers, and alopecia.
Laboratory data of the patient at the age of two (after diagnosis) and three (one year after treatment) years old.
| Parameter | 2 years | 3 years | Unit | Reference range |
|---|---|---|---|---|
| WBC | 13.8 | 6.88 | 103/μl | 4–10 |
| RBC | 4.7 | 4.68 | 106/μl | 3.9–5.8 |
| HGB | 13.1 | 13 | g/dl | 12–17 |
| HCT | 38.4 | 38.3 | % | 36–53 |
| PLT | 347 | 319 | 103/μl | 150–450 |
| Neutrophil | 30.6 | 55.2 | % | 20–50 |
| Lymphocyte | 55.3 | 36.1 | % | 50–70 |
| Monocyte | 13.7 | 8.1 | % | 2–8 |
| Eosinophil | 0.2 | 0.3 | % | 0–6 |
| Basophil | 0.2 | 0.3 | % | 0–1 |
| ESR | 17 | 12 | mm/h | 0–10 |
| BUN | 13.7 | 11 | mg/dl | |
| Creatinine | 0.6 | 0.6 | mg/dl | 0.5–1.4 |
| CPK | 122 | 52 | U/L | 40–330 |
| AST | 57 | 39 | U/L | Up to 40 |
| ALT | 101 | 36 | U/L | Up to 40 |
| ALP | 274 | 317 | U/L | 180–1200 |
| LDH | 578 | 496 | U/L | <850 |
| CRP | 1.4 | 1 | mg/L | <6 |
| ANA | 2.89 | 1.8 | index | <1.2 |
| Anti-ds DNA | 23 | 27 | U/ml | <100 |
| Anti-Phospholipid IgG | 1.2 | – | RU/ml | <12 |
| Anti-Phospholipid IgM | Neg | – | RU/ml | <12 |
| Anti-Cardiolipin IgG | 2 | 3.9 | Gplu/ml | <18 |
| Anti-Cardiolipin IgM | 2.5 | 3.2 | Mplu/ml | <18 |
| C3 | 150 | 87 | mg/dl | 90–18 |
| C4 | 27 | 17 | mg/dl | 10–40 |
| Urine protein | Neg | Neg | – | |
| Urine blood | Neg | Neg | – | |
| CSF culture | No growth after 72h | – | – | |
| CSF glucose | 70 | – | mg/dl | – |
| CSF protein | 10 | – | mg/dl | – |
WBC: white blood cell, RBC: red blood cell, HGB: hemoglobin, HCT: hematocrit, PLT: platelet, ESR: erythrocyte sedimentation rate, BUN: blood urea nitrogen, CPK: creatinine phosphokinase, AST: aspartate aminotransferase, ALT: alanine aminotransferase, ALP: alkaline phosphatase, LDH: lactate dehydrogenase, CRP: C reactive protein, ANA: anti-nuclear antibody.
She met the criteria of SLE according to the American College of Rheumatology (ACR) criteria and the diagnosis of SLE was made for the patient at the age of 2.15
TreatmentTreatment was started with methylprednisolone, hydroxychloroquine and gabapentin for the patient. For the feet spasticity, physiotherapy and hydrotherapy was started. She has had regular three months follow ups since then. The disease was under control; however, the treatment approaches had no improvement in the patient's walking disability. Abobotulinumtoxin A was injected to treat lower limb's muscle spasms in addition to multiple physiotherapy sessions, although none of them significantly improved the movement difficulties in the patient.
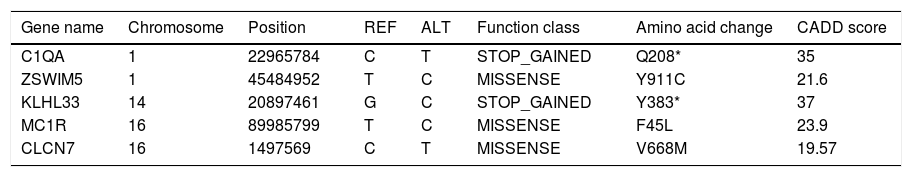
Genetic studyAnalysis of the WES from the proband identified a rare (MAF<0.001) stop-gained C>T mutation (rs121909581) in the second coding exon of C1QA gene on chromosome 1. This mutation results in changing glutamine to stop codon at amino acid position 208 (Q208*), which is predicted to be deleterious by CADD score of 35. The variant was validated in the patient using PCR and Sanger sequencing method. Segregation using the DNA sample from the family was also performed by PCR and Sanger method which showed that both parents are heterozygous for the variant while the healthy sibling was homozygous for the wildtype allele (Fig. 1). Other homozygous variants with high prediction score were not immune related and could not describe the phenotype (Table 2).
 in patient, parents and healthy sibling.")
List of homozygous rare variants (MAF<0.01) in the patient, which passed all the filtering steps.
| Gene name | Chromosome | Position | REF | ALT | Function class | Amino acid change | CADD score |
|---|---|---|---|---|---|---|---|
| C1QA | 1 | 22965784 | C | T | STOP_GAINED | Q208* | 35 |
| ZSWIM5 | 1 | 45484952 | T | C | MISSENSE | Y911C | 21.6 |
| KLHL33 | 14 | 20897461 | G | C | STOP_GAINED | Y383* | 37 |
| MC1R | 16 | 89985799 | T | C | MISSENSE | F45L | 23.9 |
| CLCN7 | 16 | 1497569 | C | T | MISSENSE | V668M | 19.57 |
ZSWIM5; Zinc Finger SWIM-Type Containing 5, KLHL33; Kelch Like Family Member 33, MC1R; Melanocortin 1 Receptor, CLCN7; Chloride Voltage-Gated Channel 7.
The C1q protein level was very low in the patient (6.9μg/ml), comparing to the normal range of 156±36μg/ml in control group.
DiscussionComplement selective deficiencies have been reported as rare genetic disorders associated with lupus-like symptoms.12 Early components of complement classical pathway such as C1Q, C2 and C4 are reported to be associated with SLE.3C1Q mutations have been reported in few populations from Turkey, Sudan, Kosovo and Iraq in SLE affected families previously. A number of these mutations have been shown to be causative for SLE.16–18
C1q has a crucial role in innate immunity, and it is believed to be important in the innate-adaptive immunity crossroad. This glycoprotein belongs to collectin family and has molecular weight of 460,000KD. It consisted of 6 heterotrimeric chains; C1qA, B and C, which is encoded by three different gene on the short arm of chromosome 1. Each chain has a collagen like N-terminal and a globular C-terminal part, considered as the recognition domain which has ligand binding capability. Full length proteins of the A, B, and C are required to produce functional C1q protein.19 Although the exact mechanism of C1q involvement in SLE pathogenesis is not known, it is believed that C1q has an anti-inflammatory function in adaptive immunity. This anti-inflammatory function is done by helping to solubilize immune complexes in addition to clearance of apoptotic debris. Therefore, in the absence of this protein, apoptotic debris accumulates and triggers autoimmunity.20
Using WES, our pSLE patient showed to have a mutant C1QA, locating in the recognition domain of the protein which is very important for normal function of C1q complex. The mutation (Q208*) creates an early stop codon which possibly disturbs not only the structure of C1qA protein but also prevents the correct assembly of the whole C1q complex as confirmed by the low serum level of protein. The genetic analysis suggests that C1q deficiency resulted in SLE symptoms in the mentioned case of the present study. It has been shown that C1q binds to apoptotic debris and accelerates the clearance of debris (auto-antigens) which is very important to maintain tolerance.21
The present investigation discloses that monogenic immunodeficiency diseases such as C1q deficiency can explain the genetic etiology of a proportion of pSLE in Iranian population. Further investigations in the population are needed to elucidate the frequency of these genetic defects among the children affected by SLE.
Making an early diagnosis of C1q deficiency is challenging, while the features could be non-classic, compared to the majority of pSLE patients. In the presented patient, pSLE started with movement disorders, which was not accompanied by any joint, kidney, or heart involvement; the frequent clinical manifestations among pSLE cases in Iran (unpublished data). Former studies explained the criteria of C1q deficient patients as malar rash (in 95% of the cases), glomerulonephritis (42%), central nervous system (CNS) involvement (18%) in addition to high titers of autoantibodies (70%).21 However, our patient had similarities and differences with the mentioned features. According to our patient's history, the following combination could be considered as C1q deficient monogenic lupus as well; early onset (below the age of five), movement disorders (with poor response to treatment), skin manifestations (i.e. malar rash), and positive ANA.
Finally, we suggest that serum C1q level should be considered as a screening test in all pSLE cases, especially in those patients with disease onset below five years old and who suffer from movement problems. C1q serum level allows early diagnosis of C1Q deficient cases, which could be confirmed later by molecular methods, i.e. PCR and Sanger sequencing of all C1Q exons and help in finding the exact mutation in such patients.
Conflict of interestThe authors have no conflict of interest to declare.
This study was supported by Tehran University of Medical Sciences, Grant No. 95-01-30-31108. We thank Dr. Zahra Aryan for sharing her comments on the clinical presentation of the case.
