


INTRODUCCION Y RECUENTO HISTORICO
La paquidermoperiostosis fue descrita por Friedreich en 1868 en dos hermanos William Hagner1 y Karl de origen germano, cuya enfermedad se inició a los doce años, ellos tenían cuatro hermanos no afectados, el padre al parecer no tenía la enfermedad y murió de inanición, la madre tenía una buena salud y se le denominó a esta enfermedad como hiperostosis (al compromiso total del esqueleto).
Erb2 y Virchow3 reexaminaron a los pacientes y describieron esta enfermedad como acromegalia, Erb observó a los hermanos durante dos décadas y le llamó la atención su «aspecto grotesco, las manos y pies gigantes y sus piernas como columna». Karl murió en 1891. Arnold4 describe la autopsia de Karl Hagner y analiza tres características de la enfermedad: acromegalia, paquidermia y la osteítis. Sternberg5 en 1899 revisó nuevamente a los dos hermanos Hagner y a pesar de que los presentó en la New Sydenhan Society como acromegalia, él pensaba que era una entidad ósea nueva. Otra familia fue descrita también en Alemania por Fraentzel6 en 1888 y él también la denominó «on acromegaly». Pierre-Marie en 1890 y Gourand7 en 1889 son los primeros franceses en describir la enfermedad y son los responsables del apodo que se les decía a los que padecían esta afección como «grosses-pattes» (giant-paws). En 1889 Gourand7 presentó al paciente a la sociedad médica de París y la denominó «grosse-pattes» pero el paciente de 50 años no había tenido ninguna molestia, Tournier en 1891 informa su caso y lo describe así: «Maladie hypertrophiante singulière: lésions élephantiasignes des parties molles et du squelette». La misma descripción realiza Oehme en 19198.
En 1935 Touraine y Solete9,10 son los primeros en realizar una descripción comprensiva en dos artículos y distinguen tres formas: una forma completa con paquidermia y con paquiperiostosis; una forma incompleta sin compromiso del cráneo y una forma frustra en la que existe un compromiso de la cara y del cráneo, pero no se observa los cambios, a nivel de los huesos especialmente en el periostio y si existen son mínimos o ausentes en el momento del examen. Vague en una extensa revisión publicó en dos artículos en 1948 y 1950 un informe sobre 60 pacientes de la Literatura y sus casos están de acuerdo con esa clasificación11,12.
Previamente los dermatólogos de la segunda escuela Vienesa como Jadassohn13 describe en 1906 el engrosamiento de la piel a nivel del cráneo y de la piel de los huesos frontales, sus pliegues tortuosos y la denominó cutis verticis gyrata, Unna14 un año después realiza la misma descripción.
Desde la primera descripción por Friedreich1 en 1868 se encuentra la asociación familiar de los hermanos Hagner, y el padre y la hija descrita por Fraentzel6 como «Über Akromegalie» en 1888, Leva15 en 1915 informa dos casos de acromegalia familiar en dos primos, y esta relación se describió como una «Ring of consanguinity», Oehme8 en 1919 describe cuatro familiares y define la asociación como «familial acromegaly like disorder, especially of the skeleton». Brugsch16 en 1941 fue el primero en reconocer la naturaleza familiar de la enfermedad y en 1950 Franceschetti et al17 informan sobre 15 casos, todos con historia familiar y plantean que la enfermedad se puede heredar en forma recesiva o denominante pero con variabilidad en la expresión clínica y con una predisposición al sexo masculino. La enfermedad se ha descrito en varias razas, especialmente en la mayoría de los informes los pacientes son de raza blanca, pero se ha descrito también en los japoneses, negros africanos, africanos-americanos, hindúes y en Latinoamérica en Perú, México y Colombia.
Touraine, Solente y Golé en 19359,10 fueron los primeros en reconocer y diferenciaron esta enfermedad de la acromegalia y de la osteoartropatía hipertrófica de origen pulmonar. En 1951 Findlay y Oosthuizen18 acordaron denominar a la enfermedad como paquidermoperiostosis, y la personificaron como el síndrome de Touraine-Solente-Golé, como se le reconoce hoy día, se desconoció la descripción de Friedreich1 y de Arnold4 como debería denominarse esta enfermedad.
A partir de la descripción de Touraine, Solente y Golé9,10 se describieron varios informes algunos relacionados con los aspectos óseos19,20, otros enfatizan los aspectos cutáneos21-23 y otros artículos la asociación del compromiso óseo y las alteraciones cutáneas24-26. El nombre paquidermoperiostosis define estrictamente la enfermedad y lo utilizó por primera vez Seze y Jurmand en 195027, Findlay et al en 195118, Shawarby et al en 195328 Angel en 195729, Cosalli y Biella en 196230, Vogl et al en 196231 con esta denominación es como se conoce la enfermedad.
A pesar de que en la osteoartropatía hipertrófica pulmonar las manifestaciones articulares son frecuentes, el compromiso articular en la paquidermoperiostosis no es frecuente, los informes en los cuales se asocie a una poliartritis son pocos, Matlingly32 fue el primero en informar que la poliartritis podría ser más una forma de presentación de la paquidermoperiostosis. La asociación con otras displasias esqueléticas es rara, solo Fam33 en 1983 describe un caso de paquidermoperiostosis displasia epifisaria múltiple y osteoartritis en 1983.
La displasia diafisaria progresiva es una enfermedad rara, generalizada, que compromete en forma simétrica los huesos largos, ocasionando un engrosamiento cortical de la diáfisis de los huesos, en ocasiones puede afectar la metáfisis, pero respetando la epífisis de los huesos. El primer caso fue informado por Cockayne34 en 1920, dos años después Mario Camurati informa otro caso35, Guido Engelmann36 en 1929 informa otro caso con las mismas características clínicas y radiológicas de los casos descritos previamente. Neuhauser et al37 en 1948 propuso el nombre de displasia diafisaria progresiva en vez de enfermedad de Engelmann, ya que la enfermedad debería llamarse Cockayne-Camurati.
Camurati34 en su artículo informa la presencia de la enfermedad en hombres de cuatro generaciones sucesivas, pero fue Girdany38 en 1959 quien describe seis pacientes de ambos sexos y demuestra una herencia autosómica dominante. Generalmente la enfermedad se inicia en la niñez y puede progresar en la adolescencia; el compromiso óseo casi siempre es bilateral, y afecta especialmente los huesos largos como fémur, tibia, peroné, húmero, cúbito y radio en ese orden. También se compromete la reja costal, los huesos de la mano y pies, además de la base del cráneo. Los síntomas más frecuentes son dolor en los miembros inferiores, debilidad muscular, dificultad para correr, fatiga fácil y cefalea. Las pruebas de los laboratorios que suelen alterarse son: la sedimentación globular acelerada, los estudios de laboratorio sobre metabolismo óseo son normales. Los signos físicos son: huesos largos, engrosados, con pérdida de modelamiento, pobre masa muscular, marcha difícil y exoftalmos.
Por primera vez en la literatura médica Walker39 en 1964 describe dos pacientes a quien se le demuestra una sobreposición de cuatro enfermedades de tipo distrófico y esclerosante. Posteriormente Michael Whyte et al40 en 1981 exponen un caso y revisan la literatura y describen cuatro variantes. Informamos sobre un paciente de 30 años, raza mestiza, que desde los dos años tiene una serie de alteraciones radiológicas de paquidermoperiostosis, displasia diafisaria (Engelmann-Camurati) pero que los criterios de toda la signología clínica y radiológica de las dos patologías mencionadas anteriormente, por lo que planteamos la posibilidad de una distrofia esquelética mixta no esclerosante, cuya expresión ósea ha sido incompleta o frustrada de las dos patologías descritas previamente como el síndrome de Tourainte-Solente Golé y la displasia diafisaria tipo Engelmann-Camurati, no descrita en la literatura médica.
CASO CLÍNICO
Se trata de un paciente de 30 años, de raza mestiza, natural de la zona rural de Cundinamarca y procedente de Bogotá, quien consulta por presentar una masa hemiescrotal derecha de cuatro años de evolución con un crecimiento lento y asintomático.
ENFERMEDAD ACTUAL
La enfermedad se le inició a los dos años por dolor y aumento de volumen especialmente de los tobillos, episodios que se repitieron cíclicamente y que le ocasionaban incapacidad, estos episodios de oligoartritis mejoraban con reposo y antiinflamatorios no esteroideos, en algunas ocasiones al paciente fue necesario hospitalizarlo. Estos episodios se presentaban en forma monocíclica cada tres o cuatro años siempre con las mismas características, en algunos de estos episodios presentó dolores en las muñecas. Durante su infancia se pensó en una artritis reumatoidea juvenil seronegativa de tipo oligoarticular de inicio temprano. A partir de los seis años notó un aumento desproporcionado de las manos, muñecas, pies y tobillos que le impedía utilizar calzado de ningún tipo dado el gran tamaño de sus tobillos y de sus pies. Notó además un crecimiento desproporcionado de sus rodillas y de sus extremidades superiores e inferiores en general, durante esta edad el paciente presentó episodios de oligoartritis especialmente de tobillos, rodillas, muñecas y ocasionalmente en codos, que mejoraban con antiinflamatorios.
EXAMEN FISICO
El paciente se encuentra en buenas condiciones generales, hemodinámicamente estable. Tensión arterial de 130/80, pulso:74, frecuencia respiratoria por minuto: 18.
El examen cardio-pulmonar: normal y el abdomen: no presentaba megalias.
Al realizar la inspección genital se encuentra una masa en hemiescroto derecho de más o menos 0,5 cm de diámetro, dura, no adherida a los planos testiculares y sin adenomegalias satélites.
APARATO LOCOMOTOR
En cuanto al aparato locomotor se observa a nivel de la piel de la cara un engrosamiento de la piel, con aspecto arrugado, que le impedía un pellizcamiento (cutis verticis girata), arcos ciliares prominentes, aumento de volumen de las clavículas, pero estas características de la piel no se observaron en los miembros superiores o inferiores.
MANOS
En las manos aparece acropaquia, macrodactilia e hipocratismo digital pero no se encontró sinovitis.
PIES
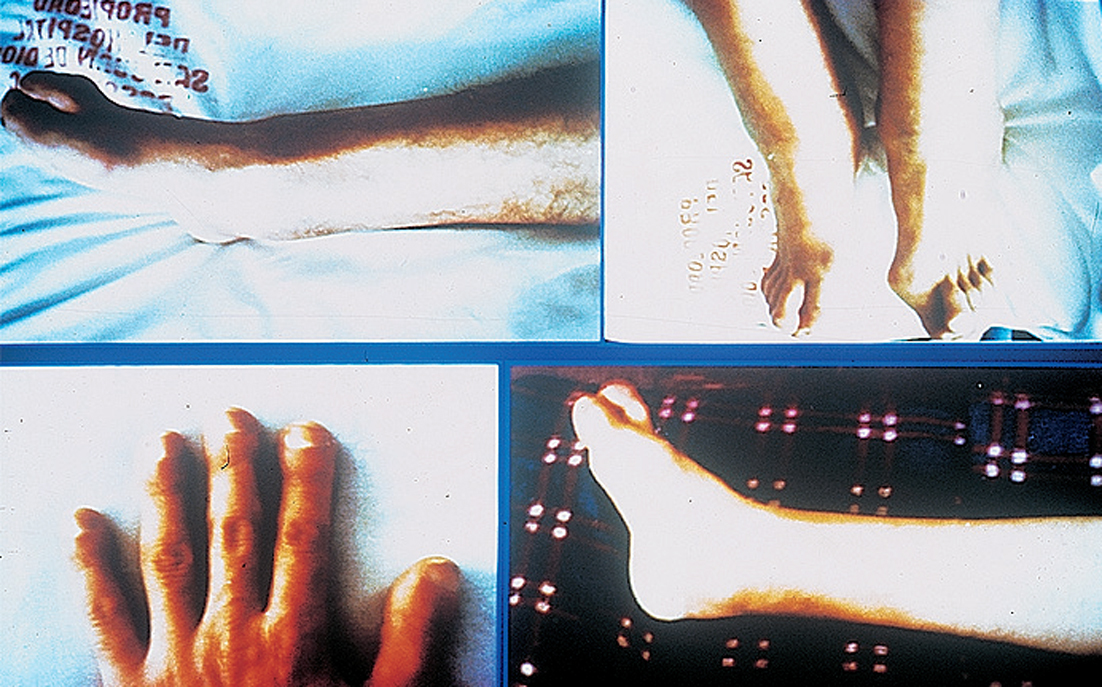
Aparece aumento del diámetro de los tobillos, macrodactilia e hipocratismo digital (fig. 1).
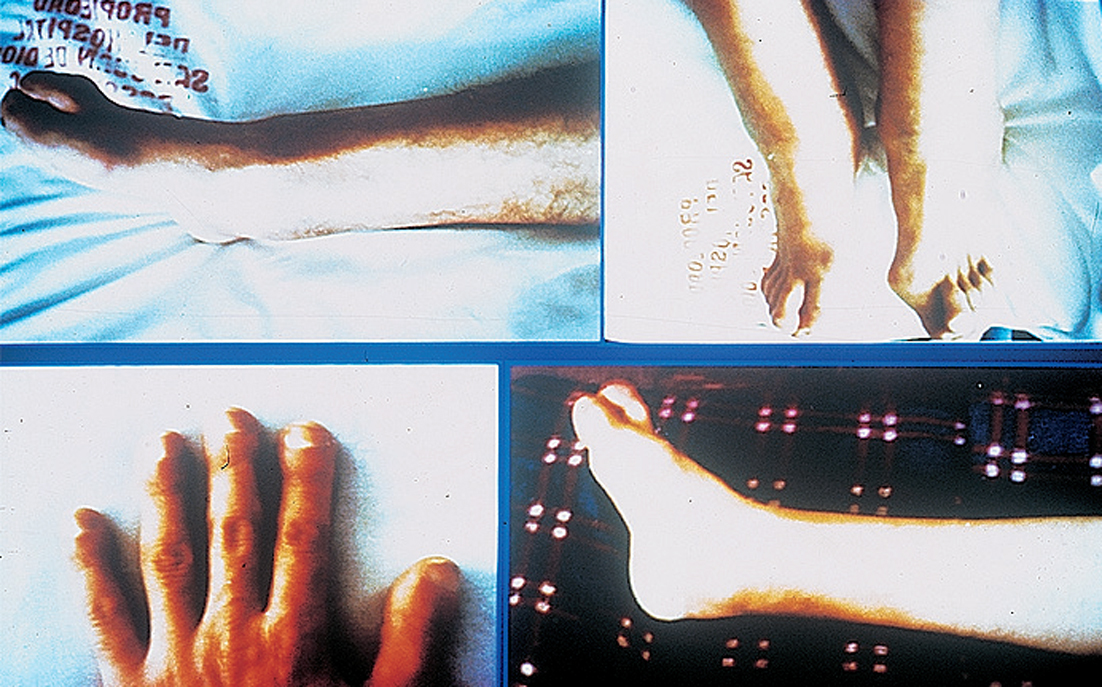
Figura 1.
HUESOS LARGOS
Se observa una hipertrofia de los miembros superiores e inferiores pero se le notó una disminución de la masa muscular y una hipotrofia del tejido celular subcutáneo (fig. 1).
ESTUDIOS DE IMAGENES
Se realizó radiografía de fémur, tibia, peroné, radio y cúbito.
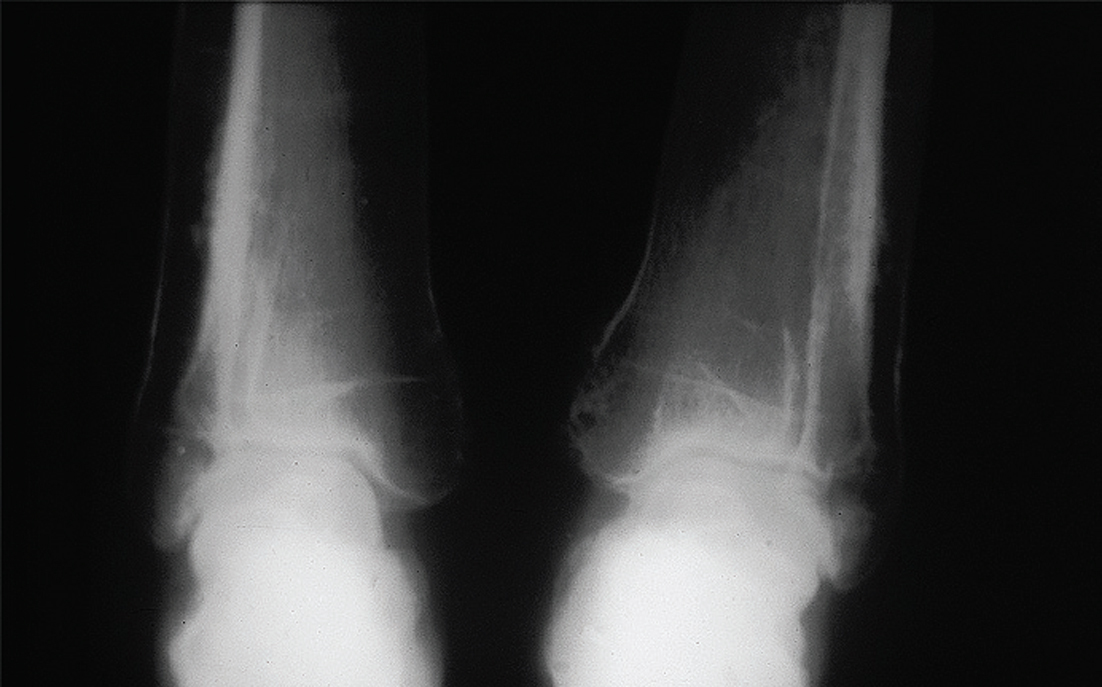
Se observa un engrosamiento fusiforme de la diáfisis de todos los huesos largos, incluyendo los huesos de las manos y pies. No se observa una hiperostosis diafisaria marcada como se observa en la hiperostosis cortical generalizada, en la enfermedad de Van Buchem41 y la enfermedad de Ribbing42. No se observan alteraciones a nivel del periostio como se observa en la osteoartropatía hipertrófica o en la paquidermioperiostosis. Sólo se observó una discreta alteración del periostio a nivel del primer tercio inferior de ambas tibias. El compromiso fusiforme de los huesos largos es simétrico. Este tipo de alteración es compatible con una displasia diafisaria (fig. 2).

Figura 2.
A nivel de los huesos del cráneo se observa una esclerosis de la base del cráneo y del maxilar inferior, pero discreta.
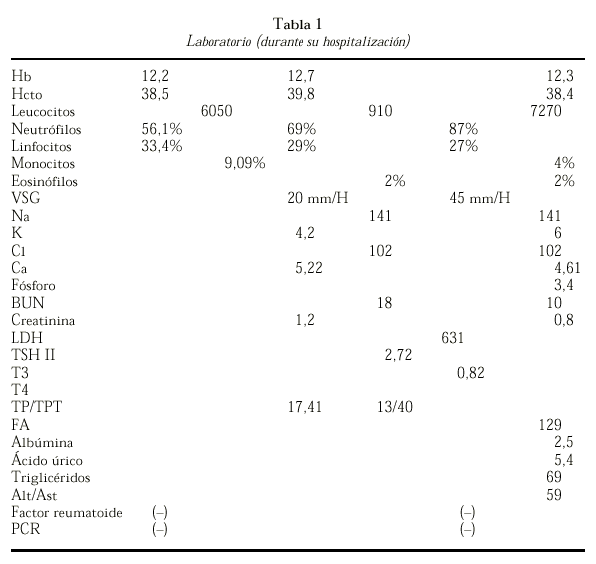
LABORATORIO
Se le practicó biopsia sinovial compatible con una sinovitis crónica inespecífica y biopsia a nivel de la masa testicular compatible con un seminoma. Asimismo se le practicó escisión de la masa.
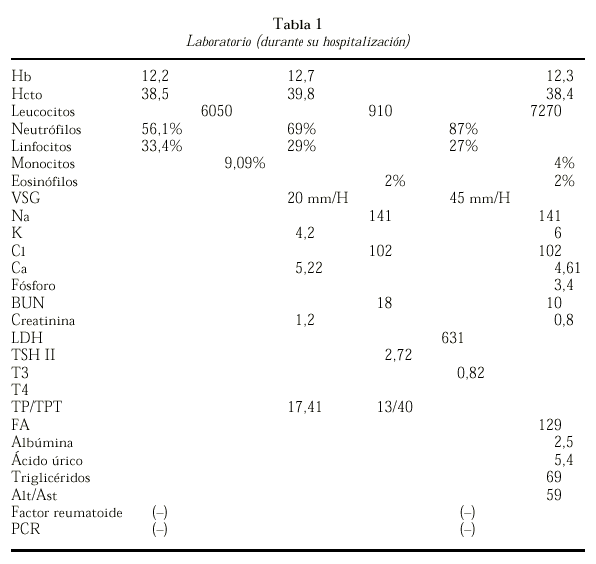
Se le practicó una resonancia magnética de los huesos largos donde se observa un aumento del diámetro de los huesos largos sin incremento del periostio, se aprecia palidez de la médula ósea (fig. 3 y tabla 1).
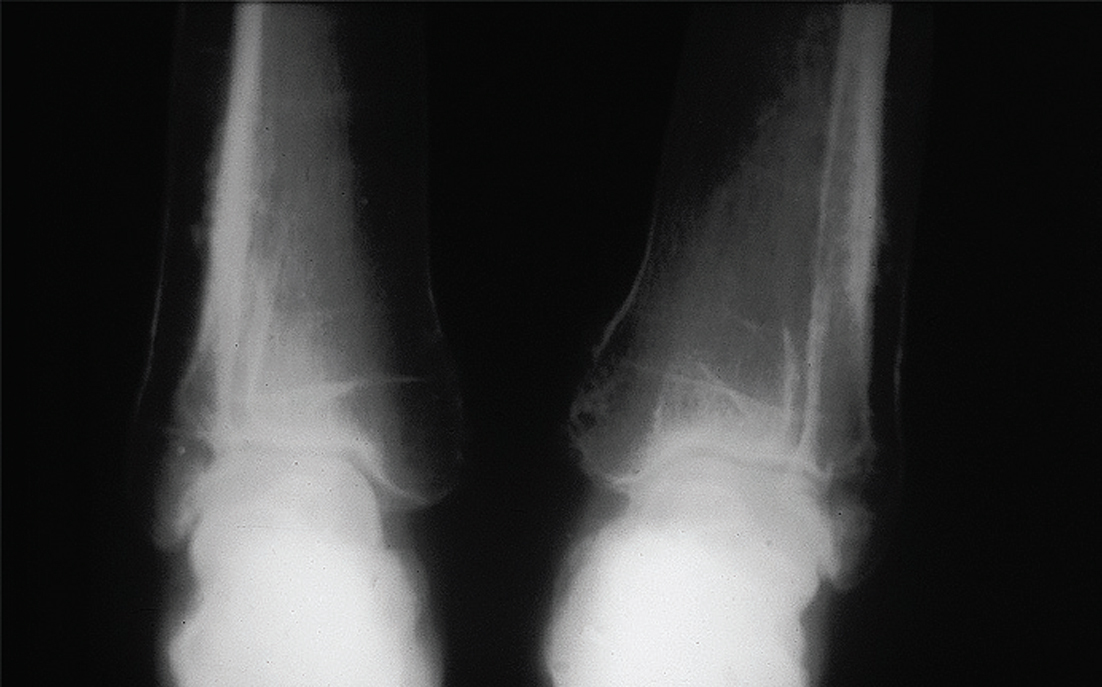
Figura 3.
COMENTARIOS
Llamaba la atención la dificultad en la marcha del paciente, por el tamaño de sus miembros (macrosomía), la poca masa muscular y el aumento de volumen de los tobillos. Además se observa a nivel de la piel de la cara una forma frustra de cutis verticis gyrata, acropaquia de las manos y pies y un hipocratismo digital desde los 18 años compatible con una forma incompleta de paquidermoperiostosis o síndrome de Touraine-Solente-Golé9,10. Al revisar los estudios radiológicos simple y de resonancia magnética de los huesos largos no compatibles con una paquidermoperiostosis sino con una displasia diafisaria tipo Engelmann-Camurati pero incompleta. La historia clínica del paciente y de los estudios radiológicos están de acuerdo con una sobreposición de dos enfermedades óseas no esclerosante.
DISCUSION
Las enfermedades óseas que comprometan la diáfisis de los huesos y el periostio crean confusiones en el diagnóstico, debido a las dificultades a nivel de las clasificaciones morfológicas de las enfermedades metabólicas óseas y de las displasias esclerosantes óseas, especialmente si compromete la diáfisis en forma simétrica o asimétrica, el tipo de herencia (autosómica dominante o recesiva) y el grado de penetrancia.
El compromiso de la diáfisis de los huesos especialmente si éste es simétrico o si la penetrancia suele ser leve que ocasiona un aumento de volumen y produce un trastorno en el modelamiento de los huesos, puede inducir errores diagnósticos, debido en ocasiones a la clasificación morfológica de este tipo de patología específica. Además no se ha logrado documentar una alteración metabólica ósea ni se ha logrado demostrar un defecto estructural a nivel del excesivo crecimiento óseo que ocasiona un trastorno en el modelamiento óseo tampoco se ha logrado demostrar alguna alteración en aquellos informes donde se demuestra la posibilidad de tener varias patologías óseas en un solo individuo, debido a que las publicaciones son escasas y generalmente son reportes de casos, por lo raro de estas patologías.
Al revisar los artículos donde se describe la esclerosis diafisaria hereditaria múltiple por Ribbing en 1949, en la que el autor define como el compromiso simétrico del fémur y la tibia con esclerosis diafisaria «diferente a la enfermedad de Engelmann-Carumati, al estudiar y observar este artículo y otros, sobre la enfermedad de Ribbing, realmente lo que apreciamos es una hiperostosis endosteal generalmente localizada y sin defectos en el modelado óseo, diferente a la enfermedad de Engelmann-Camurati que tie ne un efecto en el modelado óseo en miembros inferiores y además compromete la base del cráneo, lo que nos permite cuestionar la existencia de la enfermedad de Ribbing43 como tal. Esta confusión nos ha permitido revisar la problemática de las hiperostosis cortical, las periostosis, las displasias esqueléticas con hiperostosis y periostosis, especialmente este caso que no reunía los criterios para establecer el diagnóstico de paquidermoperiostosis ni el de enfermedad de Engelmann-Camurati, sino que tenía criterios de las dos patologías.
El solapamiento o sobreposición de varias enfermedades en un individuo está bien identificada en las enfermedades del tejido conjuntivo como la enfermedad mixta del tejido conjuntivo, la sobreposición de lupus y artritis reumatoidea, la sobreposición de esclerodermia y polimiositis, pero el solapamiento de varias enfermedades óseas en un solo individuo es bastante raro.
A partir de 1964 Walker39 informó sobre dos pacientes que tenían la sobreposición o solapamiento de una osteopoiquilia, osteopatía estriada y meloreostosis en un solo individuo; él denominó esta agrupación de distrofias óseas como «mixed sclerosing bone dystrophies» (distrofias óseas esclerosantes mixtas). Cada una de estas entidades poseen una anormalidad radiológica característica, la causa y la patogénesis no está bien definida. Posiblemente su origen sea un defecto en la osteogénesis del hueso trabecular que podría ser una alteración de tipo vascular, un defecto en la embriogénesis o una alteración del tejido colágeno. Otros investigadores como Abrahamson44 informa acerca de un caso con las mismas características de los dos pacientes de Walker en 196839; casos similares han sido publicados por Kanis y Thomson en 197545, Elkeles en 197646, Whyte et al en 198140, Kessler et al47 en 1983. Pascaud et al48 describen la asociación de melorreostosis, osteopoiquilia y esclerodermia lineal en 1981, Greenspan et al43 describen la asociación de la displasia mixta esclerosante ósea con una displasia epifisaria hemimélica (o enfermedad de Trevor-Fairbank).
En la descripción del caso de Whyte et al en 198140 al revisar la literatura sobre la sobreposición de distrofias óseas esclerosantes los autores observaron osteopatía estriada y esclerosis craneal, osteopoiquilia asociada a melorreostosis, por lo que Whyte et al40 proponen cuatro tipos de sobreposición de distrofias óseas esclerosantes de la siguiente manera: una forma es la asociación de osteopatía estriada, melorreostosis, osteopoiquilia y osteoesclerosis focal, otra es la asociación de osteopatía estriada y esclerosis focal con y sin osteopoiquilia, una tercera asociación es la osteopatía estriada con hiperostosis cortical generalizada y ensanchamiento diafisi-metafisiario con o sin esclerosis craneal y osteopoiquilia de las costillas, la cuarta asociación observada es la de osteoporquilia con proliferación de la diáfisis y el periostio. Esta clasificación de Whyte40 en nuestro criterio es una descripción morfológica y en todas está incluida la osteopoiquilia. Además, estas distrofias óseas pueden asociarse a una serie de alteraciones vasculares como la linfangiectasias unilaterales, hemangiomas capilares, malformaciones arteriovenosas y la enfermedad de Trevor (o displasia epifisaria hemimélica). Estas distrofias óseas de acuerdo a la clasificación de las oestocondro-distrofias publicada por Spranger en 199249 hacen parte de las displasias con incremento de la densidad ósea, pero esta clasificación es más morfológica, por lo que nuestro grupo está más de acuerdo con la propuesta de Greenspan en 199150 en la que se plantea la posibilidad de un defecto en la osificación endocondrol que compromete en forma secundaria al hueso maduro y así de esta forma el origen de las distrofias ósea mixtas es un defecto de la osificación ósea del hueso maduro.
Las patologías óseas de tipo distrófico y displásicas que afecten el periostio, la diáfisis, la metáfisis y además que se asocian con esclerosis e hiperostosis son poco frecuentes, pero es aún más claro la asociación de una displasia diafisaria tipo Engelmann-Camurati con paquidermoperiostosis como el presente caso, y lo más raro es que no encontramos descrita esta asociación. Lo que nos llamó la atención es el inicio de la enfermedad a los dos años como una artritis reumatoidea juvenil y posteriormente a los seis años notó aumento desproporcionado de las manos, pies y de las extremidades, que le dificultaba la marcha tipo «grosses-pattes» como lo describió Pierre-Marie51 u Gourand7 pero con hipotrofia muscular. Desde el punto de vista radiológico era una displasia diafisaria, pero desde el punto de vista clínico se comportaba como una paquidermoperiostosis. A partir de los 15 años hasta los 18 años se expresó fenotípicamente el aspecto cerebroide de la cara, como lo denominaban las tribus peruanas el «chajrahuma» como lo describió Marroquín52, pero que inicialmente fue descrita por Jadassohn13, Unna14, Stühmer53 y Sisson54 como cutis verticis girata y la acropaquia1,2 pero ambas afecciones eran leves. Estos dos aspectos son de la paquidermoperiostosis y no se observan en la displasia-diafisaria, pero los aspectos cutáneos en las extremidades no eran característicos de la enfermedad de Touraine-Solenté-Gole9,10, sino que la hipotrofia muscular está asociada a la displasia diafisaria tipo Engelman-Camurati34-37. Sólo se observó periostosis en los huesos a nivel del primer tercio inferior de la tibia y peroné, pero también el compromiso articular se hizo especialmente a nivel de los tobillos. El seminoma es otro hallazgo asociado que no tiene ninguna importancia con respecto a la enfermedad ósea. Planteamos que es el primer caso de sobreposición o solapamiento de dos enfermedades óseas como la paquidermoperiostosis y la displasia diafisaria tipo Engelmann-Camurati, y que su origen se debe a un defecto en el origen membranoso de los huesos, y que se presenta en una forma autosómica recesiva, ya que el caso de Fam et al33 informado en 1983 es la descripción de una mujer de 56 años, cuyas fotografías clínicas y radiológicas son características de una paquidermoperiostosis, la displasia epifisaria múltiple no se observa claramente en el material radiológico mostrado.
NOTICIAS
V CONGRESO INTERNACIONAL DE LA SOCIEDAD IBEROAMERICANA DE OSTEOLOGÍA Y METABOLISMO MINERAL (SIBOMM)
Noviembre 29-Diciembre 1, 2000 Buenos Aires (Argentina)
Presidente Comité Organizador Local
Dr. Carlos A. Mautalén
e-mail: sibomm@arnet.com.ar
Sede
Hotel Sheraton Libertador
Secretaría Técnica
MCI
Viamonte, 965, 6° A
1053 Buenos Aires. Argentina
Tel: 5411 4325-1273/1290
Fax: 5411 4326-8517
e-mail: mci@sion.com.ar