



Tectona grandis es un árbol maderable de importancia económica en bosques tropicales y subtropicales. Mediante este estudio, se identificaron familias de factores de transcripción (FTs) y genes codificantes para enzima, diferencialmente expresados en el xilema del tallo, implicados en la regulación de la respuesta a estrés abiótico y xilogénesis en T. grandis. Así, fue analizada la distribución evolutiva de 19 genes codificantes para FTs de T. grandis mediante análisis filogenéticos. También, fue utilizada la minería de bases de datos y publicaciones para identificar 320 genes de Arabidopsis thaliana (ortólogos a T. grandis) como soporte experimental y predictivo. Como resultados, se encontraron FTs de las familias bZIP, MYB, NAC, ER, bHLH, NuY y genes que codifican enzimas. Así mismo, se logró analizar el interactoma de T. grandis encontrando correlaciones de Pearson significativas para genes que regulan vías metabólicas de fenilpropanoides y estrés abiótico. Además, la red de coexpresión reveló nodos y aristas entre los genes TgRAP1, TgMyB1, TgHSF1, TgMyB3, TgNAC1, TgTsiid1, TgLieTFs1, TgNuy3, TgRAP2 y TgNuy4. En particular, los análisis de ontología génica mostraron 31 genes de respuesta a estrés abiótico, principalmente TgHShT1, TgHSF1 y TgHSF2 como correguladores. Además, se encontró que el regulador maestro TgNAC1, está involucrado en la corregulación de otros factores de transcripción.
Teak (Tectona grandis) is a timber tree of economic importance in tropical and subtropical forests. The aim of this work was to identify families of transcription factors (TFs) and enzyme-coding genes differentially expressed (DREs) in stem xylem and their regulation involved in abiotic stress response and xylogenesis in T. grandis. Therefore, the evolutionary distribution of 19 TFs of T. grandis was derived using a phylogenetic analysis. Besides, specific data mining procedures of databases and publications were performed in order to identify 320 Arabidopsis thaliana genes (orthologous to T. grandis) as experimental and predictive support. As results, we found transcription factors of the bZIP, MYB, NAC, ER, bHLH families, and enzyme-coding genes. Furthermore, interactome analysis in T. grandis showed a significant Pearson correlation for genes regulating metabolic pathways of phenylpropanoids and abiotic stress. Also, the coexpression network revealed nodes and edges between TgRAP1, TgMyB1, TgHSF1, TgMyB3, TgNAC1, TgTsiid1, TgLieTFs1, TgNuy3, TgRAP2 and TgNuy4 genes. Gene ontology analyses showed that 31 genes respond to abiotic stress, mainly TgHShT1, TgHSF1 and TgHSF2, as co-regulators. In addition, the TFs master regulator TgNAC1 was found to be involved in the co-regulation of other TFs.
La Teca (Tectona grandis) es un árbol caducifolio de bosques tropicales y subtropicales, que posee un rápido crecimiento y es apreciado por su madera (Anish et al., 2015). Además, esta especie tiene un gran potencial para captura de carbono e incrementar su biomasa para formación de energía renovable y sustentable (Krause et al., 2006; Quiala et al., 2012). A pesar de los numerosos estudios fenotípicos en T. grandis, poco se conoce de la especie a un nivel molecular y bioquímico (Diningrat et al., 2015; Galeano et al., 2014). Así, la tecnología de RNAseq (secuenciación de RNA) permite revelar genes diferencialmente expresados (DERs) (Wan et al., 2012) y dentro de ellos, identificar algunos factores de transcripción (FTs) implicados en respuestas fisiológicas a diferentes factores ambientales o cambios metabólicos (Diningrat et al., 2015; Galeano et al., 2015). Algunos procesos que podrían ser regulados por este tipo de factores de transcripción son el depósito de celulosa, hemicelusosa y lignina en la pared celular. Este tipo de genes reguladores pueden activarse por variación en la concentración de sales en la célula, estrés hídrico, deficiencia de nutrientes, oxidación, alta osmolaridad o cambios de condiciones de luz y temperaturas extremas (Gill & Tuteja, 2010; Prasch & Sonnewald, 2013).
Generalmente, los FTs reconocen a los promotores corriente arriba (del inglés, upstream) en las secuencias de genes que serán regulados y pueden ser activados por procesos biológicos y ambientales (Matsui et al., 2008). Además, se han observado interacciones de algunas familias de FTs para dar respuesta fisiológica a diferentes tipos de estrés y a procesos metabólicos secundarios, como el depósito de lignina y posterior producción de madera (Lin et al., 2015; Sundar et al., 2008). Los factores de transcripción son proteínas que al unirse al DNA pueden interactuar con otros reguladores transcripcionales (como pueden ser los reguladores maestros), en procesos como remodelación de la cromatina, modificación de proteínas, reclutamiento o bloqueo de las RNA polimerasas que acceden a la cadena de DNA para el proceso de transcripción (Udvardi et al., 2007).
Por otro lado, la tecnología de RNAseq es una técnica cuantitativa que ayuda a determinar niveles de expresión de RNA y su aplicación directa puede direccionarse a la construcción de bases de datos a gran escala para hacer estudios de redes de coexpresión (Dameron et al., 2013; Mizrachi et al., 2010). Dentro de las largas listas de genes detectadas por el RNAseq en diferentes condiciones ambientales, de edad o de tejidos, se pueden analizar por bioinformática patrones de expresión similares entre sí (también llamado coexpresión); genes con patrones y funciones biológicas similares pueden ser anotados por ontologías génicas y luego ser agrupados (Consortium, 2000; Dameron et al., 2013). Además, la sistematización y agrupación de los genes coexpresados, pueden estar apoyados con resultados experimentales (Consortium, 2000; Dameron et al., 2013), y de ese modo poder elucidar rutas moleculares que rigen a los organismos vivos (Fröhlich et al., 2007; Jin et al., 2014). Actualmente, se conocen 58 familias de FTs en plantas (Jin et al., 2014; Naika et al., 2013), de las cuales algunas de ellas cumplen funciones importantes en la regulación de la expresión génica, especialmente al inicio de la transcripción. Por ejemplo, la familia bZIP, caracterizada por poseer un zíper de leucinas, y la familia bHLH, distinguida por tener dos hélices alfa unidos como dímeros al DNA, se encuentran en todos los organismos eucariontes, regulando procesos fisiológicos y de desarrollo central (Ariel et al., 2007; Sauvé et al., 2004). Así, el objetivo de este trabajo bioinformático fue identificar familias de factores de transcripción (FTs) y genes enzimáticos diferencialmente expresados (DERs) en el xilema del tallo de Teca y de los que deriven posibles implicados en la regulación a la respuesta de estrés abiótico. Finalmente, es importante analizar, sistematizar e integrar los diversos datos de las redes de coexpresión génica y la información generada de diferentes experimentos de expresión génica.
MATERIALES Y MÉTODOSSelección de genes de interés diferencialmente expresados en el xilema secundario del tallo de Tectona grandisLa búsqueda y selección de genes enzimáticos de interés relacionados con la respuesta a estrés abiótico y la regulación de las vías metabólicas de síntesis del xilema secundario del tallo, fue realizado a partir del transcriptoma de T. grandis, el cual está depositado en el “Transcriptome Shotgun Assembly” (TSA número de acceso GDLT00000000), disponible en el NCBI (www.ncbi.nlm.nih.gov), obtenido por Galeano y colaboradores en el año 2015 (Galeano et al., 2015). De esta base de datos, fueron descargados 2,413 genes diferencialmente expresados. Las funciones de estos genes fueron posteriormente anotadas mediante el software Blast2Go (https://www.blast2go.com/blast2go-pro).
Identificación de factores de transcripción en T. grandis e identificación de ortólogos en Arabidopsis thalianaUn total de 19 factores de transcripción de tipo basales, activadores, coactivadores y reguladores maestros, fueron seleccionados manualmente a partir de las anotaciones realizadas con el Blast2Go a los genes diferencialmente expresados de Teca. Posteriormente, la secuencia codificante de los 19 FTs de Teca fue utilizada como referencia en las bases de datos “TAIR” (http://www.arabidopsis.org/) y “plant TFTDB” (http://planttfdb.cbi.pku.edu.cn/), para seleccionar los FTs ortólogos en A. thaliana que tuvieran un porcentaje de identidad mayor a un 70%.
Minería de literatura (Text mining)Fue realizada una búsqueda de artículos relacionados con factores de transcripción y genes enzimáticos de A. thaliana ortólogos a los genes de Teca, que estuvieran implicados experimentalmente en la regulación de las vías metabólicas relacionadas con defensa de plantas, respuestas a estrés y síntesis de xilema secundario. Para ello, se utilizó la herramienta basada en internet para minería de literatura denominada PubTator (www.PubTator/index.cgi) (Wei et al., 2012). Así, en esta búsqueda avanzada se utilizaron como palabras clave los códigos de los genes y proteínas establecidos por el NCBI. Además, se utilizaron los descriptores biológicos “gen ortólogo”, “factor de transcripción” y “genética vegetal” como filtros para disminuir el número de publicaciones. Asimismo, fueron rastreadas bases de datos en A. thaliana con relación a redes de coexpresión (http://atted.jp/) y respuesta de estrés abiótico (http://caps.ncbs.res.in/stifdb/browse.html#genename). Se utilizaron las bases de datos de Arabidopsis por ser una planta modelo, con numerosos recursos bioinformáticos disponibles.
Dendrogramas y dominios conservados de los factores de transcripción diferencialmente expresados de T. grandisLa identificación de las secuencias codificantes para proteína (del inglés Coding sequence, o CDS) fueron obtenidas con el uso del programa ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi) y la traducción de nucleótidos a aminoácidos del CDS fue realizado con el programa ExPasy (http://web.expasy.org/translate/). Posteriormente, la búsqueda de similitud proteica y de dominios se realizó con la base de datos de pFam (http://pfam.xfam.org/). De los 19 factores de transcripción identificados entre los genes diferencialmente expresados, se seleccionaron de 4 a 8 genes ortólogos con porcentaje de identidad mayor a 70% correspondientes a cada FTs. Luego, se realizaron alineamientos múltiples para cada FTs con el programa Clustal Omega y edición de secuencias mediante el programa Bioedit. Los alineamientos fueron almacenados en formato “.aln” y posteriormente transformados al formato “.meg” con el uso del software MEGA6, versión 6.06 (http://www.megasoftware.net/mega.php). Finalmente, con este programa se obtuvo un dendrograma según el método de Neighbor Joining.
Análisis de redes de interacción entre A. thaliana y T. grandisPara obtener la red de interacciones (tipo coexpresión, interacciones físicas, predicciones y dominios de proteína compartidos) de los factores de transcripción de T. grandis, se utilizaron como referencia los 19 factores de transcripción de A. thaliana ortólogos a Teca. Para obtener el interactoma de Teca, se utilizó el complemento Genemania en el software Cytoscape (http://www.cytoscape.org/), el cual utiliza la información disponible sobre la regulación génica de A. thaliana (como especie modelo) para predecir y ajustar las gráficas de interacción de otras especies de plantas, de las cuales no se conoce su regulación. Genemania utiliza el principio de redes de peso (del inglés weight networks) para ponderar las predicciones y calcular el coeficiente de correlación de Pearson para cada par de genes.
Enriquecimiento funcional basado en categorías de ontología génicaEl análisis de enriquecimiento funcional basado en categorías de ontología génica (del inglés, Gene Ontology) de los 19 FTs y 301 genes enzimáticos de A. thaliana fue realizado mediante la herramienta web de ontología génica atriGO (http://bioinfo.cau.edu.cn/agriGO/) y el complemento “Bingo” del software Cytoscape (http://www.BiNGO/Home.html). El sondeo de enriquecimiento por minería de datos se realizó a un valor p de corte ≤ 0,01 después de aplicar la corrección Benjamini - Hochberg.
RESULTADOSGenes diferencialmente expresados obtenidos del xilema secundario del tallo de T. grandis y minería de literaturaSe seleccionaron 19 FTs de T. grandis que pertenecen a 13 familias, incluyendo bHLH, MyB, HSF, NAC, Mad box, bZIP, ARF, ERF, NY, IIIBTFs, IIETfs, gata zinc, Tsiid (Archivos adicionales 1) y 301 genes enzimáticos (Archivos adicionales 2), en donde los genes ortólogos comparten regiones de dominio funcional por encima del 70% de similitud. Por otro lado, se encontraron 1,641 publicaciones relacionadas con investigaciones de respuesta a factores de estrés usando como modelo A. thaliana depositados en PubTator. Posteriormente, se obtuvieron 1,683 FTs distribuidos en 19 familias relacionados con estrés biótico y abiótico, regulando la expresión de 4,172 genes con funciones de señalización de ácido abscísico, estrés por frío y sequía, respuesta a la luz, variaciones en las concentraciones de sales celulares, estrés oxidativo y rehidratación, entre otros (Sowdhamini et al., 2009). Trabajos recientes en árboles como Populus trichocarpa y Pinus taeda han aumentado la información sobre las familias de factores de transcripción inmersos en procesos de respuesta a estrés abiótico y de biosíntesis de madera.
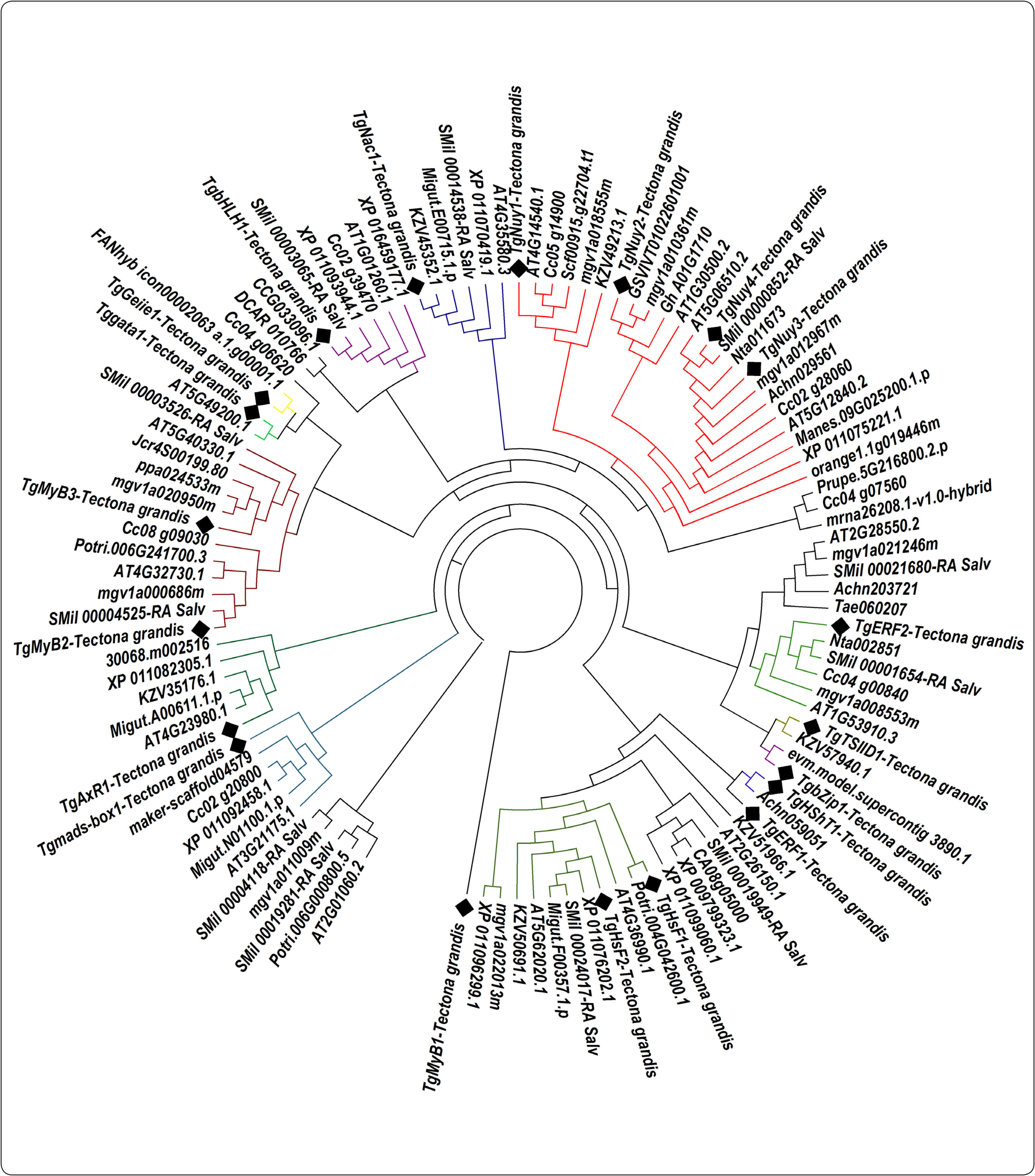
Dendrograma para la determinación de familias y motivos conservados de FTs de T. grandisLos FTs de Teca con ortólogos en A. thaliana poseen dominios funcionales involucrados en la regulación de la expresión génica, y con procesos de estrés abiótico (Tabla I). Así, la familia de FTs HSF, clases A y B, es una de las familias encontradas más abundantes que posee el motivo de localización nuclear y mantiene la conformación de giro de hélice para lograr la interacción con el DNA (Harrison et al., 1994). Este dominio sirve para regular la respuesta al calor y otros tipos de estrés ambiental, siendo la planta perenne Mimulus guttatus la más próxima a T. grandis en motivos conservados (Figura 1).
Motivos conservados evolutivamente de 18 factores de transcripción de T. grandis, pertenecientes a las familias MyB, HSF, HShT, ERF, bZIP, NAC, bHLH, Gata, Nuy, IIETFs y ARF
| FTs Tectona grandis | Motivos Conservados |
|---|---|
| Tg MyB1 | QITEALKLQMEVQKRLHEQLEVQRQLQLRIDAQGKYLKKIIEEQQ |
| Tg MyB2 | IVKGPWSKEED |
| Tg MyB3 | DQEEDLIIRLHKLLGNRWSLIA |
| Tg HSF1 | FKHNNFSSFVRQLNTYGFKK |
| Tg HSF2 | FARDLLPKYFKHNNFSSFVRQLNTYGFRKVVPDRWEFSND |
| Tg HShT1 | KRRLT |
| Tg ERF1 | KEEFVHILRRQSTGFSRGSSKYRGVTLHKCGRWEARMGQFLGKK |
| Tg ERF2 | RKRKNQYRGIRQRPWGKWAAEIRDP |
| Tg bZIP1 | SNRESARRSRLRKQKHLD |
| Tg NAC1 | EWFFFCPRDRKYPNG |
| Tg bHLH1 | LNHVEAERQRREKLNQRFYALRAVVPNISKMDKASLLGDA |
| Tg Gata1 | LCNACGLMWANKG |
| Tg Nuy1 | REQDRFLPIANVSRIMKKALPANAKISKDAKETVQECVSEFISFITGEASDKCQREKRKTINGDDLLWAMTTLGFE |
| Tg Nuy2 | SRHLHAMRRPRGNGGRFL |
| Tg Nuy3 | RAKAELEKK |
| Tg Nuy4 | RKPYLHESRHLHA |
| Tg IIETFs1 | DEYFHCE |
| Tg ARF1 | RGQPRRHLLTTGWSTFVTSKRL |
, son representados con los cuadrados de color negro. Los clúster representan las asociaciones de secuencias de aminoácidos de FTs con un porcentaje de similitud superior al 70%. El clado con más FTs es constituido por los FTs TgNuy1, TgNuy2, TgNuy3 y TgNuy4, la familia MyB y HSF tienen 3 factores de transcripción, del mismo modo son detalladas las demás familias de genes reguladores en Tectona grandis.")
Árbol filogenético de las familias de los factores de transcripción diferencialmente expresados en xilema secundario de T. grandis, (bHLH, MyB, HSF, NAC, Mad box, bZIP, ARF, ERF, NY, IIIBTFs, IIETfs, gata zinc y Tsiid), son representados con los cuadrados de color negro. Los clúster representan las asociaciones de secuencias de aminoácidos de FTs con un porcentaje de similitud superior al 70%. El clado con más FTs es constituido por los FTs TgNuy1, TgNuy2, TgNuy3 y TgNuy4, la familia MyB y HSF tienen 3 factores de transcripción, del mismo modo son detalladas las demás familias de genes reguladores en Tectona grandis.
Dentro de las familias HSF, RAP, ARF, bHLH, MyB, NF-Y (FTs que regulan la síntesis de pared celular y estrés abiótico), los genes más relevantes fueron TgMyB1, TgMyB2, TgMyB3, TgHSF1, TgERF1, TgERF2, TgNuy1, TgNuy2 y TgNuy3.
El gen TgMyB1 correspondió con Myb-like de M. guttatus (mgv1a011009m) y P. trichocarpa (Potri.006G000800.5) (Figura 1), todos ellos conteniendo la secuencia conservada LHEQLE (Tabla I). El gen TgMyB2 correspondió con Myb-like de M. guttatus (mgv1a000686m) y P. trichocarpa (Potri.006G241700.3), TgMyB3 con Myb-like de M. guttatus (mgv1a020950m), Jatropha curcas (Jcr4S00199.80) y Prunus persica (ppa024533m) (Figura 1). TgHSF1, el cual posee dominios con interacción al DNA (Tabla I) mostró homología con M. guttatus (mgv1a022013m), P. trichocarpa (Potri.004G042600.1) y A. thaliana (AT4G36990.1). También, el gen TgERF1, caracterizado por regiones de aminoácidos que reconocen la secuencia GCCGCC en plantas, está relacionado con M. guttatus (mgv1a021246m) y T. aesticum (Tae060207), y el FTs TgERF2 con M. guttatus (mgv1a008553m) y Nicotiana tabacum (Nta002851). Además, la familia NF-Y presentó para el gen TgNuy1, homología con Utricularia gibba (Scf00915.g22704.t1) y M. guttatus (mgv1a018555m), TgNuy2 con M. guttatus (mgv1a010361m) y Vitis vinifera (GSVIVT01022601001), TgNuy3 con N. tabacum (Nta011673) y M. guttatus (mgv1a012967m).
Asimismo, analizando los dominios conservados de la familia de factores de transcripción MYB, se observa que los genes TgMyB2 y TgMyB3 mantienen el dominio de unión al DNA (Tabla I), teniendo patrones estructurales tipo hélice-giro-hélice y tres triptófanos para la formación de un núcleo hidrofóbico (Ogata et al., 1995). Por otro lado, las regiones conservadas del gen TgHSF1 (familia HSF, o Heat shock) poseen dominios de interacción proteína–proteína (Figura 2), también tipo hélice-giro-hélice (Kotak et al., 2004). Además, el gen TgHSF2 posee un dominio de interacción al DNA, indicando una mayor especificidad al DNA (Tabla I). El gen TgERF2 (de la familia ethylene response factor) posee un dominio rico en arginina relacionado con la unión al esqueleto azúcar-fosfato (Allen et al., 1998). También fueron encontrados los FTs TgbZIP1 (familia bZIP), TgbHLH1 (familia bHLH), factores de transcripción basales que mantienen un zíper de leucinas y de reconocimiento de promotores corriente arriba y corriente abajo, donde el dominio de TgbHLH1 se une a los promotores formando dímeros (Schumacher et al., 2000). Asimismo, fueron identificados los genes TgNuy1,TgNuy2, TgNuy3, TgNuy4 ricos en Gln, Ser, Thr y pertenecientes a la familia NF-YA, con dominios de interacción en el surco menor de DNA y el motivo CCAAT (Nardini et al., 2013). Finalmente, los dominios conservados de la familia de factores de transcripción NAC se caracterizan por poseer un dominio conservado NAC N-terminal. En este estudio, fue encontrado el gen TgNAC1 (Tabla I). Los genes de esta familia pueden interactuar y regular con otros FTs y al mismo tiempo unirse al DNA, no poseen motivos hélice-giro-hélice y generalmente la estructura del monómero de dominio es β-hoja antiparalela y doblada (Nole-Wilson & Krizek, 2000). Para los dominios conservados del gen TgARF1 se ha observado que los dímeros del motivo ARF se unen como pinzas moleculares y de reconocimiento específico en los promotores corriente arriba o corriente abajo (Boer et al., 2014).

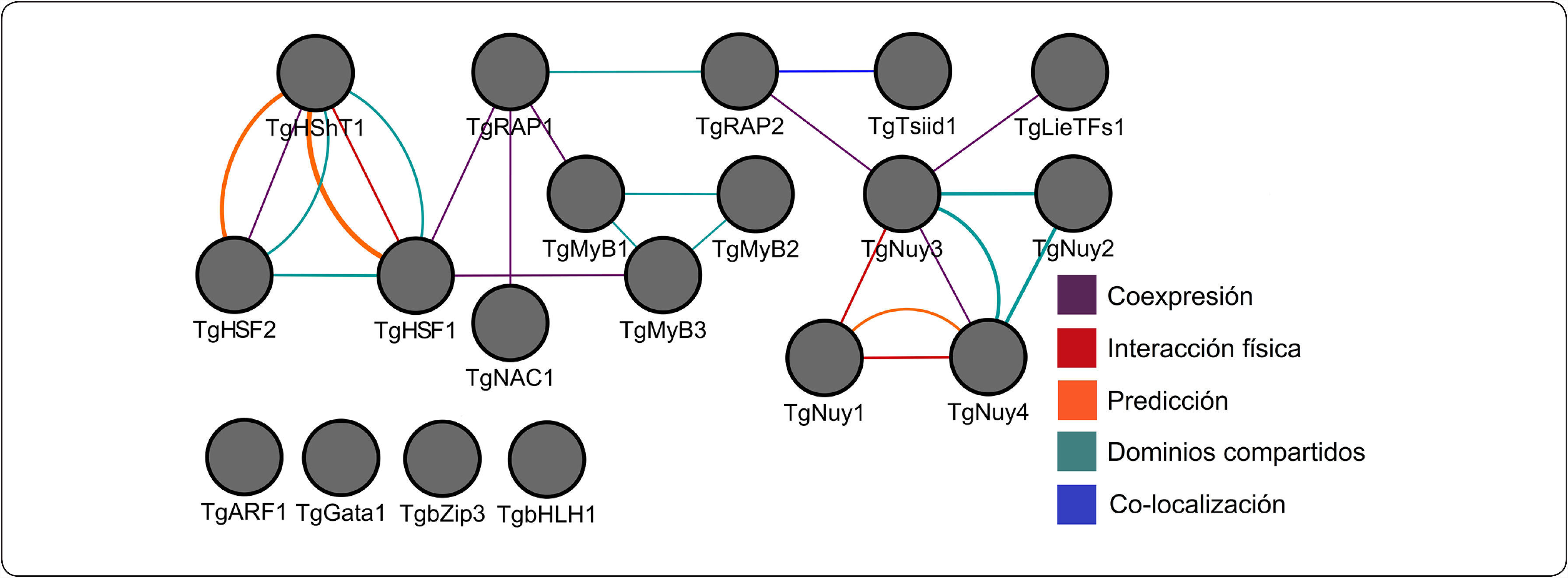
Interactoma de 19 FTs diferencialmente expresados en tejido xilemático de tallo en T. grandis. Se incluye la red de coexpresión, donde los FTs TgRAP1, TgRAP2, TgMyB1, TgHSF1, TgHShT1, TgHSF2, TgMyB3, TgNAC1, TgLieTFs1, TgNuy3 y TgNuy4 se interconectan para hacer emerger propiedades funcionales de las células. Asimismo, se muestran interacciones físicas entre los FTs TgHShT1-TgHSF1 y TgNuy3-TgNuy4-TgNuy1. Además, se presentan diferentes redes como predicciones, dominios de proteína compartidos y otros tipos de interacción.
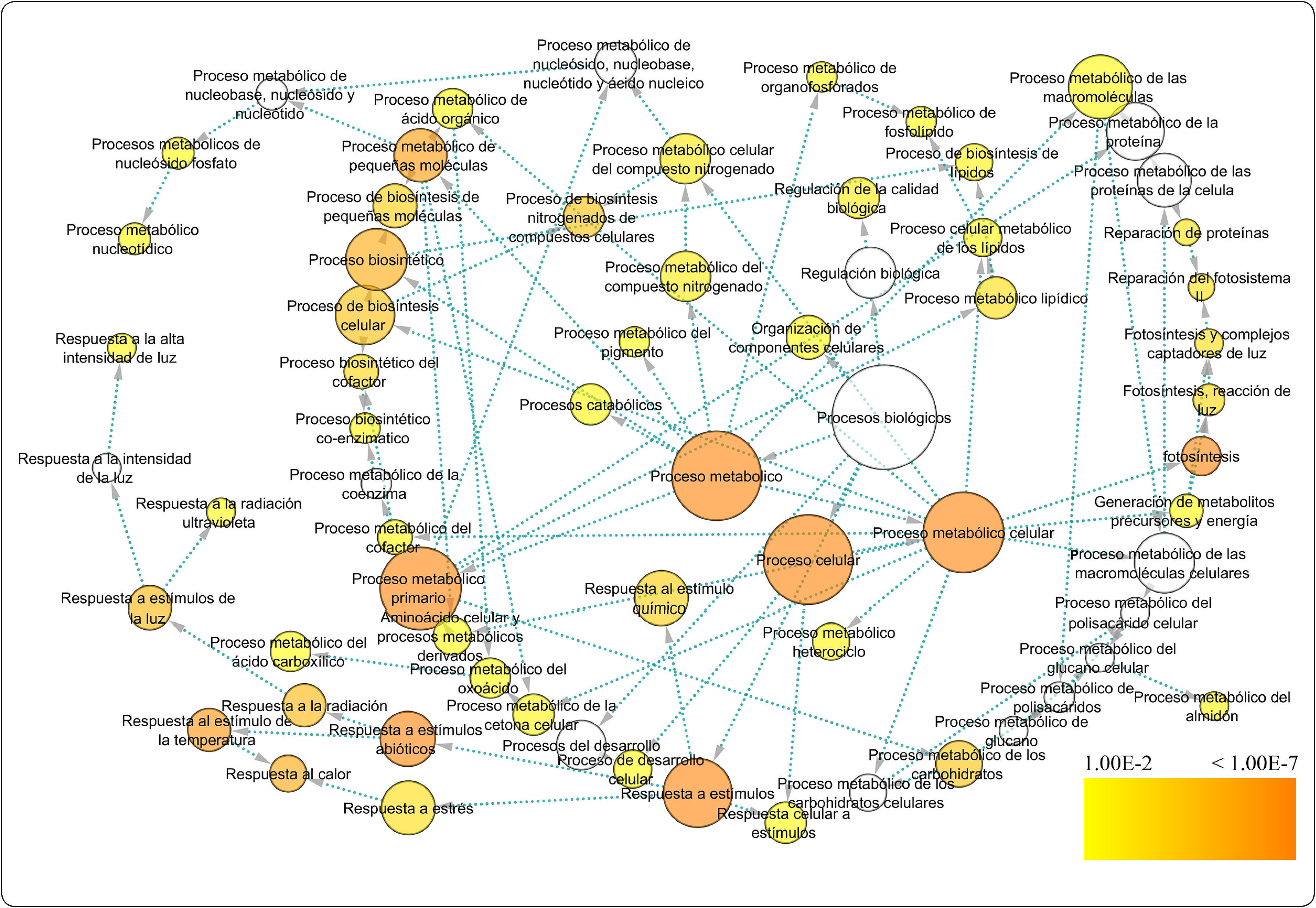
Los genes de A. thaliana en la red de coexpresión se dividen en grupos de acuerdo a su función (Figura 3), (Tabla II). De este modo, se identificaron genes relacionados a la respuesta de estrés abiótico. Se seleccionaron 301 genes enzimáticos y 19 FTs con un porcentaje de similitud superior al 70%, de los cuales 10 son de respuesta a estrés por calor, 16 responden a estrés por luz y temperatura, mientras que 108 genes están relacionados con procesos metabólicos celulares (Tabla II).

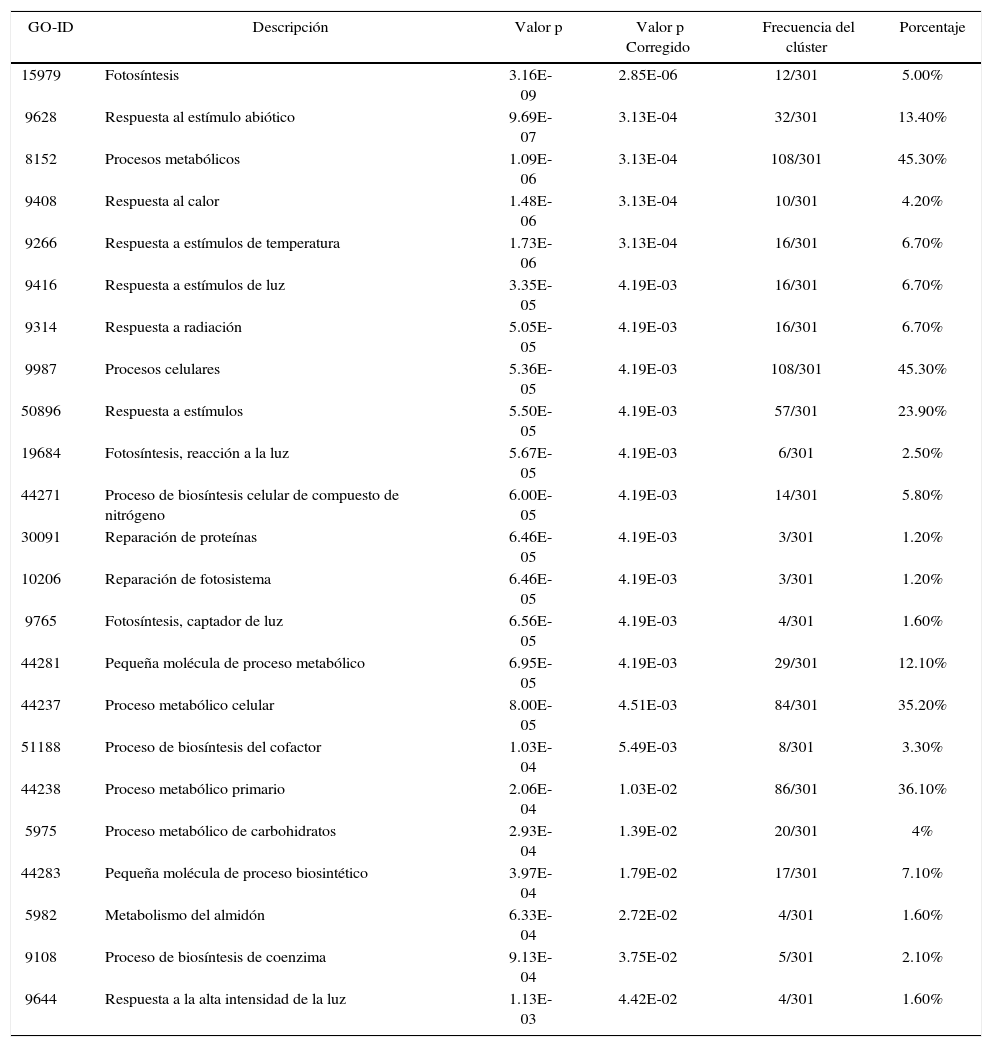
Nodos de ontologías génicas más significativos. Son incluidos sus valores p y frecuencias de clúster, donde las descripciones con mayor porcentaje son representados por las actividades metabólicas y procesos celulares
| GO-ID | Descripción | Valor p | Valor p Corregido | Frecuencia del clúster | Porcentaje |
|---|---|---|---|---|---|
| 15979 | Fotosíntesis | 3.16E-09 | 2.85E-06 | 12/301 | 5.00% |
| 9628 | Respuesta al estímulo abiótico | 9.69E-07 | 3.13E-04 | 32/301 | 13.40% |
| 8152 | Procesos metabólicos | 1.09E-06 | 3.13E-04 | 108/301 | 45.30% |
| 9408 | Respuesta al calor | 1.48E-06 | 3.13E-04 | 10/301 | 4.20% |
| 9266 | Respuesta a estímulos de temperatura | 1.73E-06 | 3.13E-04 | 16/301 | 6.70% |
| 9416 | Respuesta a estímulos de luz | 3.35E-05 | 4.19E-03 | 16/301 | 6.70% |
| 9314 | Respuesta a radiación | 5.05E-05 | 4.19E-03 | 16/301 | 6.70% |
| 9987 | Procesos celulares | 5.36E-05 | 4.19E-03 | 108/301 | 45.30% |
| 50896 | Respuesta a estímulos | 5.50E-05 | 4.19E-03 | 57/301 | 23.90% |
| 19684 | Fotosíntesis, reacción a la luz | 5.67E-05 | 4.19E-03 | 6/301 | 2.50% |
| 44271 | Proceso de biosíntesis celular de compuesto de nitrógeno | 6.00E-05 | 4.19E-03 | 14/301 | 5.80% |
| 30091 | Reparación de proteínas | 6.46E-05 | 4.19E-03 | 3/301 | 1.20% |
| 10206 | Reparación de fotosistema | 6.46E-05 | 4.19E-03 | 3/301 | 1.20% |
| 9765 | Fotosíntesis, captador de luz | 6.56E-05 | 4.19E-03 | 4/301 | 1.60% |
| 44281 | Pequeña molécula de proceso metabólico | 6.95E-05 | 4.19E-03 | 29/301 | 12.10% |
| 44237 | Proceso metabólico celular | 8.00E-05 | 4.51E-03 | 84/301 | 35.20% |
| 51188 | Proceso de biosíntesis del cofactor | 1.03E-04 | 5.49E-03 | 8/301 | 3.30% |
| 44238 | Proceso metabólico primario | 2.06E-04 | 1.03E-02 | 86/301 | 36.10% |
| 5975 | Proceso metabólico de carbohidratos | 2.93E-04 | 1.39E-02 | 20/301 | 4% |
| 44283 | Pequeña molécula de proceso biosintético | 3.97E-04 | 1.79E-02 | 17/301 | 7.10% |
| 5982 | Metabolismo del almidón | 6.33E-04 | 2.72E-02 | 4/301 | 1.60% |
| 9108 | Proceso de biosíntesis de coenzima | 9.13E-04 | 3.75E-02 | 5/301 | 2.10% |
| 9644 | Respuesta a la alta intensidad de la luz | 1.13E-03 | 4.42E-02 | 4/301 | 1.60% |
Por otro lado, los FTs TgHSF1, TgHSF2 y TgHShT1 responden a estrés de calor en T. grandis, existiendo interacciones físicas entre los nodos de TgHShT1 con TgHSF1 y TgNuy4-TgNuy3 con TgNuy1 (Figura 2). Extendiendo el modelo de predicción, se observa la interacción física de TgHSF2 con TgHShT1 y TgHSF1. La red de coexpresión entre nodos e interacción por aristas (Figura 2) están entrelazadas por los FTs TgRAP1, TgMyB1, TgHSF1, TgMyB3, TgNAC1, TgTsiid1, TgLieTFs1, TgNuy3, TgRAP2 y TgNuy4, integrando la información parcial sobre 15 FTs. Entre las familias de FTs de Teca existen 4 agrupaciones que mantienen dominios funcionales compartidos entre los genes, teniendo como primer grupo TgHSF2, TgHShT1, TgHSF, luego TgMyB1, TgMyB2, TgMyB3, siguiéndole TgNuy2, TgNuy3, TgNuy4 y finalmente TgRAP1, TgRAP2 (Figura 1) y (Figura 2).
Análisis de enriquecimiento funcional en genes de TecaLa obtención de las ontologías génicas se realizó con 19 factores de transcripción y 301 genes enzimáticos ortólogos a genes de A. thaliana involucrados en la regulación de respuesta a estrés abiótico y en la formación de pared celular. De ellos, 108 de los genes diferencialmente expresados tienen relación directa con procesos celulares y metabólicos (Figura 3) y 32 genes dan respuesta a estrés abiótico, calor, estrés oxidativo y alta intensidad de luz. Los genes TgHShT1, TgHSF1, TgHSF2 son posibles factores de transcripción de respuesta a estrés abiótico que regulan positivamente otros genes de T. grandis (Figura 2). Además, de los datos obtenidos, fueron detectados genes de FTs maestros (master regulators) que regulan el estrés abiótico en T. grandis. Entre ellos, fue encontrado el regulador master TgNaC1, involucrado en la regulación negativa de algunas funciones y ortólogo de NTL9 de A. thaliana (Yoon et al., 2008), quien responde a estrés osmótico y senescencia foliar. Asimismo, el FT TgHShT1 es ortólogo de AtHSFA2 y un regulador clave en la respuesta a calor inducida bajo estrés ambiental. El gen TgHShT1 regula la transcripción de varios genes implicados en defensa frente al estrés térmico, incrementando su transcripción rápidamente bajo señales de estrés oxidativo, y su inhibición conlleva a la disfunción de diferentes organelos y muerte celular (Liu et al., 2013; Nishizawa-Yokoi et al., 2010) (Archivos adicionales 3). Además, el gen TgHShT1 posee interacción con 14 genes ortólogos en Teca y de respuesta a estrés oxidativo (Figura 2). Asimismo, los genes TgERF1 y TgERF2 responden a procesos biológicos de crecimiento y desarrollo, así como a señales hormonales y respuesta a estrés biótico y abiótico. El gen TgMyB3 es ortólogo de MyB23 de A. thaliana, quien regula su propio promotor y está relacionado con especialización celular epidérmica y formación de tricomas (Kang et al., 2009; Kirik et al., 2001).
En la Figura 3 se muestra la comparación de un patrón de genes diferencialmente expresados de la especie modelo A. thaliana, ortólogos de T. grandis que poseen anotación biológica en funciones como respuesta a estrés abiótico tanto de estímulo de alta intensidad de luz como de respuesta a calor. Las ontologías y los genes relacionados a regulación biológica están soportados por la simulación de Benjamini y Hochberg, la cual asocia significativamente genes a determinadas funciones (i.e. respuesta a calor y alta iluminación) (Tabla II). En consecuencia, cuando se obtiene la red de coexpresión, se logra entender en mayor proporción el enriquecimiento funcional con la presencia de 320 nodos y 4,049 aristas, incluyendo genes relacionados con procesos metabólicos, fotosintéticos y de biosíntesis (Archivos adicionales 3).
DISCUSIÓNLa diversificación evolutiva de los factores de transcripción es fundamental para los procesos de adaptación de las diferentes especies frente a estímulos ambientales (Jin et al., 2014). Existen alrededor de un 45% de FTs específicos en plantas. En A. thaliana, aproximadamente un 5% del genoma se dedica a su codificación, manteniendo conservados los dominios de unión al DNA entre los miembros de cada familia de FTs (Riechmann, 2000). En este estudio, los 19 FTs pertenecientes a las familias bHLH, MyB, HSF, NAC, Mad box, bZIP, ARF, ERF, NY, IIIBTFs, IIETfs, gata zinc, Tsiid y los 301 genes enzimáticos diferencialmente expresados en xilema secundario de T. grandis cumplen funciones en procesos catabólicos, metabolismo de carbohidratos, regulación génica y respuesta enzimática al estrés biótico y abiótico (Galeano et al., 2015), producción de biopolímeros y procesos de xilogénesis. Estos FTs también fueron encontrados en los perfiles transcriptómicos de otros árboles como P. trichocarpa y E. grandis (Dharmawardhana, et al., 2010; Mizrachi et al., 2010). En particular, M. guttattus fue la especie más próxima a T. grandis, ya que mantiene dominios funcionales conservados en seis FTs ortólogos, ello debido probablemente a su cercanía taxonómica en el orden Lamiales (Scoville et al., 2012).
Por otro lado, dentro de los genes enzimáticos, se identificó el transcrito de ß-galactosidasa fundamental en la síntesis de carbohidratos, glicoproteínas y glicolípidos (Roach et al., 2011). También se encontraron genes enzimáticos como quinasas, esenciales en las vías de señalización para el crecimiento, desarrollo y percepción a las diversas respuestas de estrés ambiental en plantas (Ihnatowicz et al., 2008). Además, se encontraron genes que codifican enzimas relacionados con la actividad fotosintética y en procesos de fosforilación y activación de FTs a nivel de la membrana intracelular, regulados por reacciones químicas hormonales como el etileno y el ácido abscísico (Abe et al., 2003; Yoon et al., 2008; Zhou et al., 2014). La respuesta fisiológica de la planta a los cambios ambientales es dinámica a nivel de la expresión génica. En ese punto, los FTs maestros son seleccionados evolutivamente por las plantas para tener funciones esenciales en la corregulación de procesos metabólicos como la xilogénesis y adaptación a estrés ambiental (Banti et al., 2010; Lin et al., 2015), fenómenos que se evidencian analizando las redes de coexpresión, para describir los patrones de correlación entre los genes y lograr identificar biomarcadores (Consortium, 2000).
Así, para activar los procesos metabólicos es necesario que los motivos funcionales de los FTs interactúen físicamente con los promotores upstream para su regulación y generalmente interactúan físicamente entre ellos para regular la expresión génica (Lin et al., 2015; Obata & Fernie, 2012). Finalmente, es posible que existan diferencias en los motivos funcionales entre genes ortólogos de familias de factores de transcripción, que podrían explicar divergencias funcionales y en algunos casos se expresarían en variaciones fenotípicas (Carretero-Paulet et al., 2010; Jin et al., 2014).
CONCLUSIONESCon la información del transcriptoma de T. grandis fue posible establecer y visualizar redes de interacción de genes diferencialmente expresados en el xilema secundario del tallo, apoyados por la búsqueda de minería y bases de datos con enriquecimiento ontológico. Así, con la información disponible y utilizando el software Cytoscape (herramienta para analizar, modelar y predecir funciones génicas), se identificaron FTs implicados en procesos lignocelulósicos y respuesta a estrés abiótico.
Los patrones evolutivos de los 19 FTs analizados aquí se mantienen conservados dentro de las familias y sub familias. Además, son importantes en la regulación génica, ya que fueron identificados a partir de los genes diferencialmente expresados (DERs) en el tejido del xilema secundario del tallo. Debido a que las funciones particulares de una proteína se determinan analizando su secuencia de residuos de aminoácidos, la evaluación de dominios conservados funcionales mediante genes ortólogos de T. grandis en A. thaliana es útil para identificar posibles funciones del gen (Consortium, 2000; Dameron et al., 2013), siendo fundamental hacer uso de esta especie modelo, ya que tiene abundante recurso bioinformático disponible. También, la base de datos PubMed fue útil para obtener publicaciones que documentan evidencia experimental encontradas con el programa pubtator para filtrar la información y optimizar la sistematización de datos, la predicción de genes y el soporte experimental. Finalmente, este estudio bioinformático aumenta los estudios funcionales de genes de interés en T. grandis, árbol de gran importancia biotecnológica.
AGRADECIMIENTOSLos autores agradecen a la Escola Superior de Agricultura Luiz de Queiroz de la Universidade de São Paulo por el soporte financiero y bioinformático.