


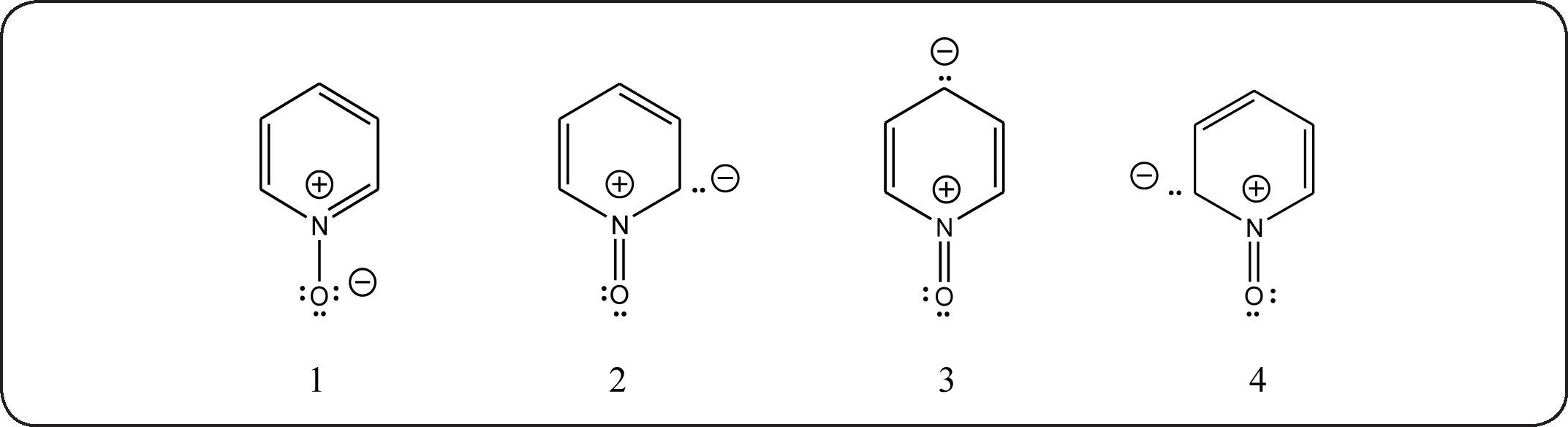
Linton encontró que el momento dipolo del N-óxido de piridina es mucho menor que el valor teórico esperado. Debido a este resultado, propuso la existencia de 3 ‘estructuras excitadas’, con carga eléctrica negativa en las posiciones 2, 4 y 6 del anillo piridínico. Sin embargo, en una reacción de sustitución electrofílica típica como lo es la nitración, solamente se obtiene el producto de reacción en la posición 4. Esta discrepancia entre lo propuesto por Linton y la reactividad observada nos indujo a estudiar las propiedades físicas del N-óxido de piridina, ya que su reactividad es resultante de las mismas. Por otra parte, las estructuras con carga negativa en el anillo requieren una polarización inesperada y hubo que proponer un mecanismo viable de formación, mediante inducción intermolecular, como se detalla en la Discusión. Además, se encontró que solamente una de las tres estructuras antes propuestas tiene soporte tanto por la reactividad observada como por datos de resonancia magnética nuclear de carbono 13. Al descartar 2 de las ‘estructuras excitadas’ de Linton, se ha explicado por qué la nitración del N-óxido de piridina es regioselectiva.
Linton found out that the dipole moment of pyridine N-oxide is appreciably smaller than the expected theoretical value. Thus, he postulated the contribution of three ‘excited structures’, with a negative electric charge at the 2-, 4- and 6-position. However, a typical electrophilic substitution such as nitration, afforded only the 4-nitro derivative. This discrepancy between theory and experiment prompted us to study the pyridine N-oxide physical properties, since its reactivity is derived from them. Besides, these negative charged rings require an unexpected polarization and a reaction mechanism must be provided. We propose intermolecular induced polarization as a viable path. We uncovered that only one of the three structures before mentioned is supported by the observed reactivity and by 13C nuclear magnetic resonance data. On rejecting 2 of the Linton’ s ‘excited structures’, we have explained the regioselectivity found in pyridine N-oxide nitration
Las propiedades químicas de un compuesto, y por ende su reactividad, derivan de su estructura molecular y de las propiedades físicas resultantes de la misma. Por lo anterior, es de primordial interés revisar la bibliografía relativa al N-óxido de piridina, ya que el conocimiento de sus propiedades físicas (propiedades esenciales1) es indispensable para poder deducir una teoría, basada en hechos experimentales, que explique el porqué de la regioquímica observada en la reacción de nitración. Ésta no ha sido explicada mas que parcialmente.
Parte Teóricaa) N-óxido de piridinaMeissenheimer2 preparó por vez primera el N-óxido de piridina al hacer reaccionar la base con ácido perbenzoico (BzO2H). Describió el picrato, el clorhidrato y el N-óxido libre, con pf 66–68°C, indicando que es delicuescente.
Años después, Linton3 realizó un estudio sobre los momentos dipolo de varios óxidos de aminas. Indicó que el N-óxido de piridina cristaliza en agujas blancas al evaporar mediante vacío una solución etérea tibia. El compuesto fue guardado en desecador sobre pentóxido de fósforo.
El momento dipolo se obtuvo determinando la polarización molar de la disolución por medición de la constante dieléctrica usando soluciones diluidas de la sustancia polar en un disolvente apolar (en este caso benceno).
El momento dipolo del N-óxido de la trimetilamina es de 5.02 D y el del N-óxido de piridina es de 4.24 D. El valor observado para la unión semipolar +N-O- es de 4.38 D. Si a éste se suma el dipolo de la piridina, el N-óxido de piridina debería tener un momento dipolo de 6.6 D, según la estructura basal, 1.
Para explicar el dipolo menor observado, se propusieron las estructuras excitadas 2 a 4, de signo opuesto, que contribuyen al estado final.
Ochiai y colaboradores llevaron a cabo una serie de investigaciones en Japón sobre la preparacióny reacciones del N-óxido de piridina y otros compuestos relacionados. Dado que los artículos estaban escritos enjaponés, Ochiai publicó en Estados Unidos una reseña de dichos trabajos4. En el apéndice de este resumen se encuentra una preparación detallada del N-óxido de piridina, empleando ácido acético glacialy peróxido de hidrógeno al 35%. El p.e. del N-óxido es de 138–140°/15mm y el rendimiento de 96%. Otros experimentos fueron descritos con anterioridad5, señalando la formación del peroxiácido intermediario.
Trabajos similares fueron llevados a cabo en Holanda por den Hertog y Overhoff6. Posteriormente se describieron las propiedades del N-óxido de piridina como donador, es decir, como ligando para formar complejos de coordinación7. Un ejemplo es el compuesto formado con el nitrato de cobalto y 6 ligandos: el hexakis derivado Co(NO3)26L, [Co(C5H5NO)6] (NO3)2
Se han determinado los espectros de absorción del N-óxido de piridina en el infrarrojo y en el ultravioleta cercano8. En su espectro infrarrojo, en solución de disulfuro de carbono, la banda en 1265cm–1 fue asignada a vibración de tensión NO (stretching). Los datos del espectro electrónico del N-óxido de piridina en solución de ciclohexano son: bandas de transición π-π* en 283mμ, log ε m=4.11 y 251mμ, log ε m=3.30.
La absorción del N-óxido de piridina en el ultravioleta cercano (295–365nm) ha sido fotografiada a alta resolución9. Los resultados indican una disminución en la distancia N-O en el estado excitado, acompañada por cambios en la estructura anular. Como puede verse esto corrobora la participación de al menos una de las estructuras propuestas por Linton.
Se determinó elespectro de microondas delN-óxido de piridina10, con el fin de establecer, por este método, el momento dipolo de esta molécula, hallando un valor de 4.13 D. Se encontró que la unión NO en el N-óxido de piridina es más corta y más fuerte que la del N-óxido de trimetilamina. Esto se debe a la deslocalización electrónica π en las estructuras resonantes con carga negativa en el núcleo. Esto mismo se reflej a en la polaridad reducida que presenta el N-óxido de piridina.
La estructura molecular del N-óxido de piridina ha sido determinada por difracción electrónica en fase gaseosa11. La distancia obtenida para la ligadura N-0 es de 1.29 Å. Esta distancia es mayor que en el óxido nítrico, NO, 1.18 Å, pero es más corta que en (CH3)3NO, 1.34 Å, con cierto carácter o. Una unión dativa N-0 es usualmente más larga que una ligadura N-O normal. Una ligadura sencilla C-N tiene un valor medio de 1.47 Å y una doble ligadura C=N tiene valores que oscilan entre 1.28 y 1.30 Å. La doble ligadura C=N en el N-óxido de piridina tiene un valor de 1.38 Å, entre simple y doble, lo cual concuerda con una estructura resonante quinoide.
Un estudio de resonancia magnética nuclear de carbono 13 del N-óxido de piridina12 reveló que la resonancia del carbono γ ocurre, comparativamente, a campo más alto (a menor frecuencia): 13C-α: 138.7; 13C-β: 125.6; 13C-γ: 123.2ppm, a campo bajo (amayorfrecuencia) conrespecto atetrametilsilano interno y en solución de tetracloruro de carbono. El corrimiento del carbono γ hacia ‘campo alto’ implica una densidad electrónica alta enesa posición, de acuerdo conuna contribución significante de la estructura semiquinoide 3 al híbrido de resonancia del compuesto en estudio.
Debido a estos resultados, en bibliografía especializada en métodos espectroscópicos13, lamolécula del N-óxido de piridina está representada precisamente por la estructura dipolar 3.
El desplazamiento químico de 15N en el N-óxido de piridina es de -86.2ppm (δ rel. CH3NO2) y DMSO como disolvente14. En la piridina el valor correspondiente es -63.2ppm y en el clorhidrato de piridina es de -164.8ppm. Cuando se utiliza ciclohexano como disolvente y el desplazamiento químico es relativo a amoniaco gas, se tiene un valor de 324.4ppm para el N-óxido de piridina15.
La estructura cristalina del N-óxido de piridina ha sido determinada por difracción de rayos X.16 Se utilizaron monocristales transparentes, ‘water white’, casi esféricos, de alrededor de 1mm de diámetro. Siendo extremadamente higroscópicos, se protegieron de la humedad mediante la presencia de un contenedor con pentóxido de fósforo colocado en los tubos sellados de Lindemann. El desecador se colocó de manera que no tocara el cristal y a su vez, no estuviera expuesto a los rayos X. Se utilizó la radiación Cu Ka. Los cristales son ortorrómbicos. Las longitudes de las uniones C-C son similares a los valores promedio observados en moléculas aromáticas. La longitud de la unión C-N es de 1.34 Å, el valor promedio en sistemas heterocíclicos conjugados. Lalongitud de launiónN-O es de 1.3 5 Å, un poco más corta que el observado en el óxido de trimetilamina (1.388 Å). De todos estos datos puede deducirse una baja contribución de las estructuras excitadas de Linton.
En otra publicación posterior de rayos X se encontró que el N-óxido de piridina forma puentes de hidrógeno intermoleculares17.
b) DiscusiónLa determinación del momento dipolo del N-óxido de piridina realizada por Linton3 hizo que éste postulara la existencia de las estructuras resonantes 2, 3, y 4, antes mencionadas.
No hay duda acerca del momento dipolo encontrado en esta molécula. La determinación del mismo mediante su espectro de microondas10 dio incluso un valor algo menor, confirmando el bajo momento dipolo en el N-óxido de piridina. Sin embargo, de las estructuras resonantes propuestas porLinton, solamente la 3 ha tenido confirmacióntanto espectroscópica como labasada en reactividad. Aunque la espectroscopía de microondas reveló que la ligadura N-O es más corta que en el N-óxido de trimetilamina, concorde con las estructuras 2,3 y 4, el espectro de resonancia magnética nuclear12 de carbono 13 encontró que solamente la señal del carbono γ (C-4) aparece a campo más alto (a menor frecuencia), no así las señales de C-2 y C-6. Esto confirma la importancia exclusiva de la estructura semiquinoide 3.
Por otra parte, de tener existencia e importancia las estructuras 2 y 4, la molécula debería reaccionar, al nitrarla, en las posiciones C-2 y C-6 para formar el N-óxido de la 2-nitropiridina, y además, en cantidad doble a la del N-óxido de la 4-nitropiridina. Sin embargo, como se detallaenel siguiente apartado, solamente hay nitración en la posición 4. Debido a la concordancia existente entre la espectroscopía de RMN y la reactividad observada, consideramos descartarlas estructuras extremas de Linton 2 y 4.
Aun cuando la estructura resonante 3 ha sido confirmada por diferentes estudios, detallados enel apartado anterior, y también por la influencia directriz del N-óxido de piridina al reaccionar exclusivamente en 4 enuna reacción de sustitución electrofílica aromática típica, la formación misma de la estructura resonante 3 presenta dificultades teóricas que no han sido contempladas y menos explicadas.
En efecto, es de sobra sabido cómo ocurre la polarización del grupo imino en la piridina. El corrimiento electrónico hacia el átomo de nitrógeno es labase de lareacciónde Tchitchibabin18,19 para obtener, por reacción nucleofílica, la 2-aminopiridina. Este corrimiento debe ser mucho mayor en el N-óxido de piridina, ya que el nitrógeno está ahora positivo (ión imonio o iminio). Sinembargo, esta tendencia de polarización debe invertirse para formar las estructuras excitadas de Linton. Se puede aducir que el oxígeno, negativo, aporta sus electrones debido a la atracción del nitrógeno positivo. Pero, no obstante este aporte electrónico que forma la doble ligadura ON, el nitrógeno sigue positivo debido a que ya tenía completo el octeto electrónico. O sea, el nitrógeno no sufrió cambio. Por otra parte, es más estable la carga negativa en oxígeno que en carbono y, además, se pierde la aromaticidad del anillo. De manera que hay que explicar por qué ocurre el corrimiento electrónico anómalo conducente a la formación de la estructura dipolar 3.
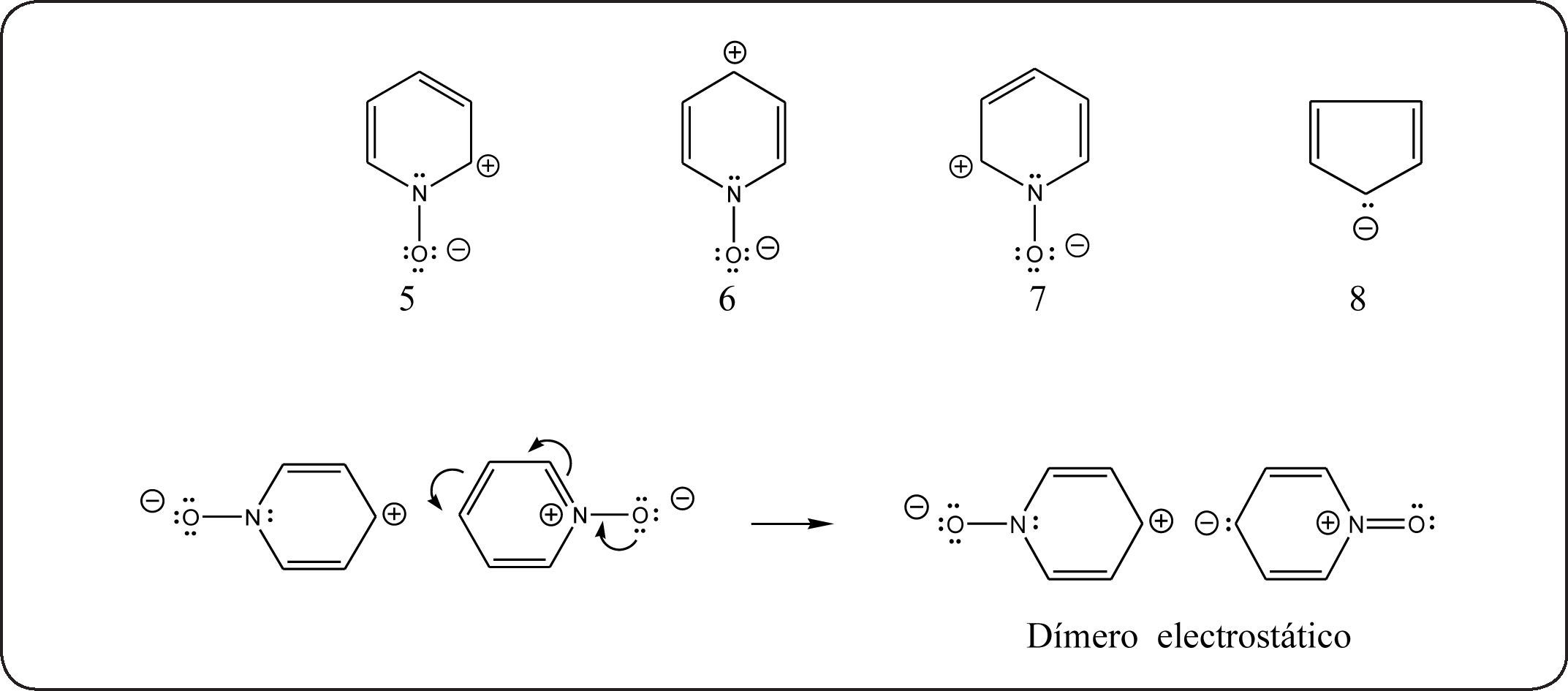
Nuestra explicación es la siguiente. El corrimiento electrónico esperado es el que forma las estructuras resonantes 5 a 7. En ellas el átomo de nitrógeno positivo es la fuerza motriz, quedando éste neutro, aun cuando esto implique la ruptura del anillo electrónico. Concomitantemente, se forman iones carbonio en 2 y 4. Estos sextetos buscan estabilizarse y una de las maneras puede ser atraer electrones de otra molécula, v. gr., como los iones nitronio. Esto sí puede inducir la polarización inesperada para formar la estructura 3 propuesta por Linton. Es decir, proponemos que el corrimiento electrónico se lleva a cabo mediante inducción intermolecular, como se indica:
Linton para explicar el momento dipolo sui generis (menor que el esperado) del N-óxido de piridina, dado que solamente24 se forma un producto de reacción.
Recientemente, la Universidad de Bremen ha descrito en forma detallada la nitración del N-óxido de piridina para obtener el ¿Por qué no se forman las estructuras 2 y 4, según indica el espectro de RMN de 13C? Obsérvese que, a diferencia de las estructuras 2 y 4, la estructura 3 tiene similitud con la del anión del ciclopentadieno, 8, de carácter aromático.
De las estructuras resonantes 5 y 6, con momentos dipolo 1,3 y 1,5, respectivamente, la 6, con un momento dipolo mayor, es la más inestable y tiende a formar el dímero electrostático propuesto.
c) Nitración del N-óxido de piridinaDado que ya era sabido20 que en la nitración del benceno la especie reactiva era el ión nitronio, Ochiai, en Japón, basado en las estructuras excitadas propuestas por Linton, llevó a cabo la nitración del N-óxido de piridina en diferentes condiciones experimentales21–23. Unos años después, den Hertogy Overhoff, en Holanda, nitraron el N-óxido de piridina6, antes de que aparecieran los resúmenes en inglés de los 2 últimos trabajos citados.
Ambos grupos de trabajo encontraron que el producto de la nitraciónes el N-óxido de la 4-nitropiridina. Como se mencionó en el apartado anterior, este resultado experimental solamente avala una de las tres estructuras excitadas propuestas por 4-nitro derivado25, y se incluyen los espectros IR, de RMN de 1H y de RMN de 13C.
d) DiscusiónSe ha señalado26 que la nitración del N-óxido de piridina en la posición 4 puede efectuarse mucho más fácilmente que la nitración de la piridina, pero que la reacción es aún más difícil que la nitración del benceno. El N-óxido de piridina se nitra con ácidos nítrico y sulfúrico concentrados, a 130°, durante 3.5 horas4, mientras que el benceno reacciona con los mismos reactivos a 50°. Cf. 27–29. La comparación resulta interesante porque nos hace ver que aún la única estructura excitada de Linton que es aceptable, la 3, no es muy importante. Es decir, a pesar de su carga negativa en la posición 4, el N-óxido de piridina tiene una reactividad más baja que el benceno, cuyo estado basal es neutro, teniendo en cuenta que la especie reactiva en la nitración es un ión positivo.
La cinética de nitración del N-óxido de piridina indica que la reacción ocurre a través de la base libre30–32. Este resultado experimental está de acuerdo con la teoría, ya que la estructura resonante 3 deriva obviamente de la base libre.
El dímero electrostático propuesto en la discusión anterior sería la fuente de la estructura resonante reactiva, el iluro 3. Sin embargo, queda por aclarar cómo ocurre la nitración del otro componente del dímero, la estructura resonante 6. Ésta debe pasar a la estructura basal original, 1, habiendo a continuación 2 posibilidades teóricas: a) Que ahora el ión nitronio genere el efecto electromérico33,34, eneste caso lapolarizacióninesperada, es decir, el corrimiento electrónico hacia el anillo, formando el electrómero35–373. El efecto electromérico ha sido llamado ‘dynamic conjugation effect’38. La otra posibilidad, b), es que solamente una parte de las estructuras resonantes 6 pase a la estructura basal y que las estructuras restantes con carga positiva en 4 generen de nuevo más estructuras resonantes 3, con carbaniones aptos para ser nitrados. Hay que hacer notar que estas alternativas teóricas no se excluyen mutuamente y pueden coexistir.
Conclusiones- •
La formación de las ‘estructuras excitadas’ propuestas por Linton para el N-óxido de piridina, con carga negativa en 2, 4 y 6, requiere una explicación teórica, ya que esto implica un corrimiento electrónico contrario al efecto mesomérico propio del grupo imonio presente en el estado basal.
- •
Para formar los iluros propuestos, el efecto electrodonador del átomo de oxígeno del N-óxido de piridina debe superar el efecto mesomérico contrario antes citado.
- •
Para que se lleve a cabo el corrimiento electrónico anómalo requerido, es necesario un excitante o promotor, es decir, debe participar el átomo positivo de otra molécula (efecto electromérico).
- •
En ausencia de reactivo, se propone la participación de la forma resonante del N-óxido de piridina que tiene carga positiva en C-4, para poder formar los iluros propuestos. Inducción intermolecular como factor operante.
- •
Las ‘estructuras excitadas’ de Linton predicen reacción del N-óxido de piridina enlas posiciones anulares 2,4 y 6. Sin embargo, en una reacciónde sustitución electrofílica típica como lo es la nitración, solamente hay reacción en C-4.
- •
La resonancia magnética nuclear de 13C del N-óxido de piridina solamente avala una densidad electrónica mayor en C-4.
- •
Estos resultados experimentales hacen descartar las ‘estructuras excitadas’ con carga negativa en C-2 y C-6, eliminando así una aporía teórica.
- •
Hemos aclarado el mecanismo de la transmisión de la resonancia en el N-óxido de piridina, al proponer la participación de inducción intermolecular. Se presentó una teoría coherente con el fin de lograr una intelección lógica y que, además, está en concordancia con los resultados experimentales, explicando la regioselectividad observada en la nitración del N-óxido de piridina.