




INTRODUCCIÓN
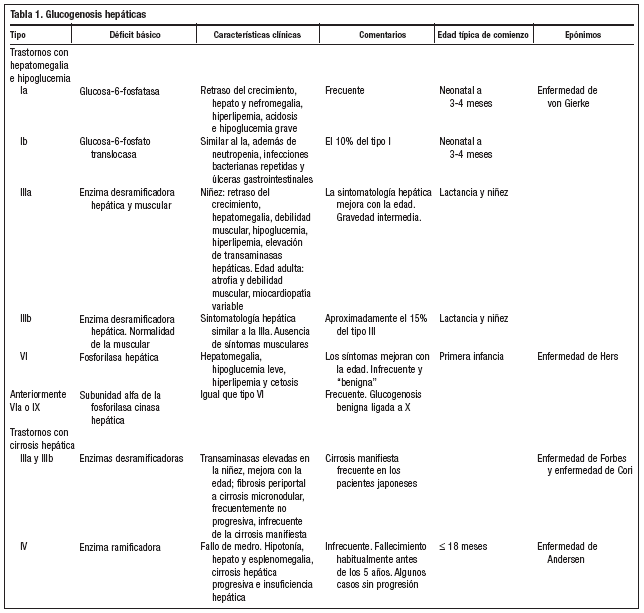
Las glucogenosis son un grupo de enfermedades hereditarias, causadas por la falta de una o más enzimas que intervienen en la síntesis o degradación del glucógeno y que se caracterizan por el depósito de cantidades o tipos anormales de glucógeno en los tejidos. Salvo el tipo VI que se transmite ligado al cromosoma X, todos los demás tipos se heredan de forma autosómica recesiva, siendo la frecuencia global de todas las formas aproximadamente de 1/20.000-25.000 nacidos vivos, aunque esta cifra podría ser demasiado baja, ya que algunas formas producen trastornos mínimos y pueden pasar inadvertidas. Las manifestaciones clínicas, la gravedad y la edad de comienzo son variables y dependen de la expresión específica de cada uno de los sistemas enzimáticos afectados de los distintos órganos1.
EXPOSICIÓN DEL CASO
Mujer de 46 años, sin alergias medicamentosas conocidas ni hábitos tóxicos, con antecedentes de mastopatía fibroquística con controles mamográficos anuales, hipoacusia crónica, depresión, insuficiencia venosa crónica, incontinencia urinaria, fibromialgia, epitelioma basocelular en el dorso de la nariz extirpado en el 2000; hipertransaminasemia diagnosticada en 1974, practicándose una biopsia hepática, con hallazgos inespecíficos, con revisiones periódicas en el servicio de medicina interna y serología de hepatitis B y C negativas. En tratamiento actual con lorazepam 1 mg/día, paroxetina 20 mg/día, cloruro de trospio 20 mg/día. Antecedentes familiares: madre con arteritis de la temporal y diabetes tipo 2; padre con hipercolesterolemia y enfermedad cardiovascular, y una de sus hermanas falleció a los 3 años de edad por una posible hepatopatía no filiada. Acude a nuestra consulta por un cuadro de cinco años de evolución, más acusado en los últimos dos años, de debilidad muscular generalizada con dificultad progresiva para la realización de sus actividades habituales. Había sufrido además varias caídas. Los síntomas no presentaban fluctuaciones a lo largo del día, ni se desencadenaban o agravaban con ningún factor conocido. En los días previos a la consulta presenta dolores musculares en piernas y antebrazos. No presentaba disfagia, diplopía, ptosis palpebral, cefalea, ni síntomas a otros niveles. No debilidad de la musculatura cervical. En la exploración física general, excepto la obesidad, no reveló otros hallazgos significativos y la exploración neurológica fue normal. Solicitamos analítica con hemograma normal, velocidad de sedimentación globular (VSG) 11 mm/h, alanina aminotransferasa (ALT) 112 U/l, aspartato aminotransferasa (AST) 107 U/l, lactatodeshidrogenasa (LDH) 918 U/l, creatinfosfocinasa (CPK) 1.294 U/l, fosfatasa alcalina y gamma-glutamil-transferasa (GGT) normales, y resto de determinaciones bioquímicas sin interés. Pedimos un electromiograma por la cifra tan alta de CPK, ante la sospecha de una enfermedad de depósito en músculo que pudiese tener relación con el hígado y justificase las cifras elevadas de transaminasas, y reveló: hallazgos de carácter inespecífico que pueden corresponder con una afectación miopática no distrófica en la musculatura proximal de extremidades. Lo remitimos al Servicio de Medicina Interna para estudio (vista primero por la consulta externa de Medicina Interna y posteriormente por Neurología): repiten analítica con hemograma normal, VSG 6 mm/h, ALT 164 U/l, AST 156 U/l, LDH 1.209 U/l, CPK 1.383 U/l, aldolasa 12,1 U/l, K 5,1 mval/l y urea 53 mg/dl, con resto de bioquímica normal. Inmunoglobulinas, factor reumatoide (FR), C3, C4, crioglobulinas y anticuerpos antinucleares (ANA) normales o negativos. Estudio electroforético (en suero) normal. Ceruloplasmina y cobre: normales. Alfa 1 antitripsina normal. Hierro y ferritina normales. Porfirinas en orina de 24 horas normales. Pruebas de función tiroidea y cortisol basal normales. Ecografía y gammagrafía abdominal: normales. Biopsia muscular: miopatía vacuolar con depósito de glucógeno. La actividad amilo 1,6 glucosidasa indetectable. Probable glucogenosis tipo III. Inician tratamiento con difenilhidantoína, pero dada la ineficacia se decide retirar e iniciar tratamiento con vitamina B. Ante la persistencia clínica le ponen carbamacepina 200 mg cada 8 horas y rehabilitación para potenciar la fuerza muscular, con mejoría clínica.
DISCUSIÓN
En los pacientes con glucogenosis tipo IIIa, la debilidad muscular suele ser mínima durante la niñez, pero puede hacerse más grave durante el tercer o cuarto decenio de la vida con debilidad lentamente progresiva y atrofia muscular. Es frecuente la hipertrofia ventricular. Los síntomas hepáticos pueden ser tan leves que el diagnóstico no se realice hasta la edad adulta, cuando se manifiesta la afectación neuromuscular, como en el caso de nuestra paciente. Se pueden utilizar los niveles séricos de CPK para identificar los pacientes con afectación muscular, pero los niveles normales no excluyen el diagnóstico. La resonancia magnética permite identificar de forma incruenta el glucógeno y algunos metabolitos intermediarios depositados en los tejidos y el diagnóstico se confirma demostrando la ausencia enzimática concreta en los tejidos afectados mediante una biopsia2,3.
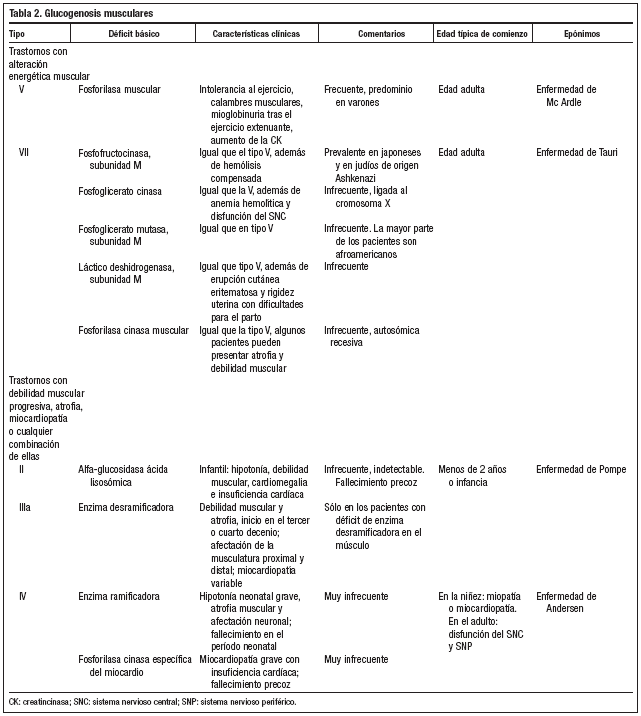
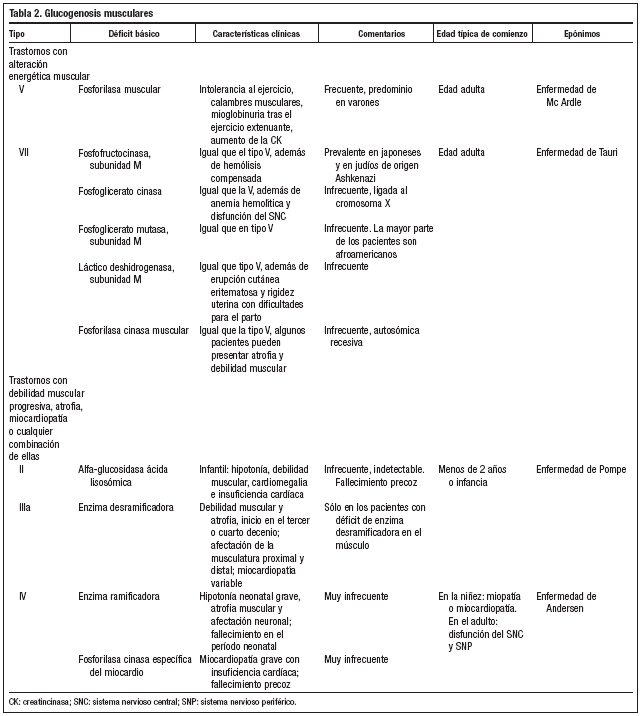
Los tejidos que se afectan con mayor frecuencia y gravedad, debido a que presentan cantidades abundantes de glucógeno, son el hígado (glucogenosis tipo I, III, IV y VI) y el músculo (tipos V y VII)2-9 (tablas 1 y 2). La glucogenosis tipo II o enfermedad de Pompe es generalizada, con importante participación neuromuscular, incluido en miocardio, siendo una de las formas más graves, que se manifiesta en el primer año de vida y suele ser mortal hacia los dos años de edad1.
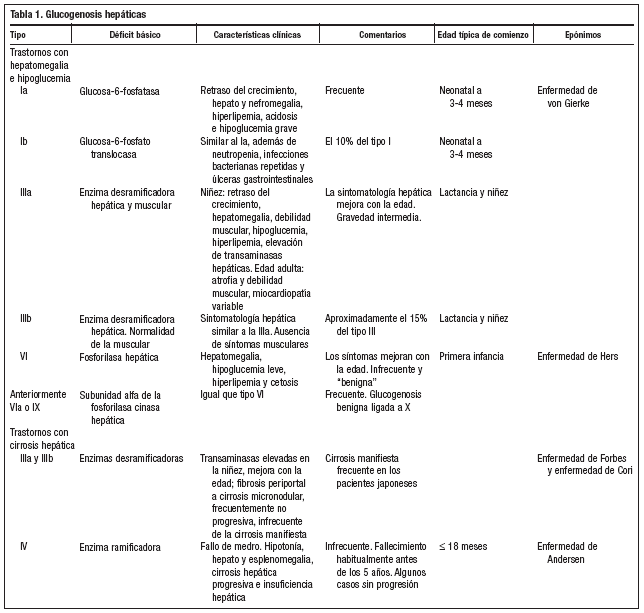
En la glucogenosis tipo IIIa la debilidad muscular y la atrofia empeoran en la edad adulta, mientras que los síntomas hepáticos mejoran con la edad y suelen desaparecer después de la pubertad2,3.
En cuanto al tratamiento de las glucogenosis que afectan predominantemente al hígado, éste va dirigido a evitar la hipoglucemia y la acidosis láctica mediante la ingesta frecuente de pequeñas cantidades de alimentos que contengan carbohidratos o con suplementos de almidón de maíz, así como la administración nocturna de alimentación mediante sonda nasogástrica; y respecto a las glucogenosis que afectan predominantemente al músculo, se tratan limitando el ejercicio anaeróbico, y en algunos pacientes resulta útil la dieta rica en proteínas4,10,11.
En el caso de nuestra paciente deberíamos hacer un diagnóstico diferencial con las distintas causas de debilidad muscular: secundaria a disfunción de la motoneurona superior (infarto cerebral, síndrome pseudobulbar, esclerosis lateral amiotrófica [ELA], espondilosis cervical, etc.), de la motoneurona inferior (poliomielitis, siringomielia, etc.), de las raíces nerviosas y nervios periféricos (hernia discal, neuropatía diabética, etc.), de la unión neuromuscular (miastenia gravis, botulismo), o del propio músculo (polimiositis, distrofias musculares, disfunción tiroidea). También deberíamos tener en cuenta las distintas causas de polialgia: reumáticas (fibromialgia, polimialgia reumática, artropatías inflamatorias, polimiositis y dermatomiositis, osteoporosis y osteomalacia), infecciosas (virus de la inmunodeficiencia humana [VIH], síndrome de fatiga crónica y otras viriasis), metabólicas (disfunción tiroidea y paratiroidea), metastásicas y psiquiátricas (trastorno de dolor persistente somatomorfo, hipocondría, trastorno de somatización, reacción adaptativa, depresión mayor, esquizofrenia, adicción a drogas, neurosis de renta, etc.). Además hemos de tener en cuenta las distintas causas de elevación de transaminasas como pueden ser: las enfermedades hepáticas (hepatitis víricas, tóxicas y medicamentosas, cirrosis, colestasis, procesos infiltrativos y tumorales), afecciones cardíacas (infarto agudo de miocardio [IAM], cateterismo, angioplastia y cirugía cardíaca, cardioversión y miocarditis aguda), afecciones musculares (traumatismos musculares extensos, convulsiones recientes, polimiositis y dermatomiositis, distrofias musculares y ejercicio muscular intenso y sostenido), fármacos (antiepilépticos, anticonceptivos orales, antiinflamatorios no esteroideos [AINE], isoniacida, tetraciclinas, inhibidores de la enzima conversora de la angiotensina [IECA], etc.), y otros procesos (cirugía reciente no cardíaca, pancreatitis aguda, alcoholismo, embolia o trombosis hística con infarto de cualquier etiología excepto cerebral, mononucleosis infecciosa y quemaduras profundas graves). Por último, recordar las distintas causas de miopatías como son: las miopatías congénitas (nemalínica), mitocondriales (oftalmoplejia externa progresiva, síndrome de Kearns-Sayre, síndrome de MELAS, síndrome de MERFF, la miopatía con distribución a nivel de las cinturas y la infantil) y las enfermedades musculares por almacenamiento de glucógeno, como es el caso aquí descrito1.
