


Membranoproliferative glomerulonephritis is a pattern of glomerular lesion with a variety of causes. It can be classified by using direct immunofluorescence into immune complex-mediated membranoproliferative glomerulonephritis and complement-mediated membranoproliferative glomerulonephritis.
ObjectiveDetermine the prevalence of membranoproliferative glomerulonephritis by biopsy (light microscopy, direct immunofluorescence and electron microscopy), categorise them according to the most recent classification, identify possible causes and determine certain epidemiological and clinical characteristics.
Material and methodA descriptive, cross-sectional study was carried out, selecting renal biopsies with a membranoproliferative pattern at 5 years at Hospital General de México. Age, gender, clinical syndrome and immunofluorescence and electron microscopy results were obtained. Biopsies lacking immunofluorescence or electron microscopy results were excluded.
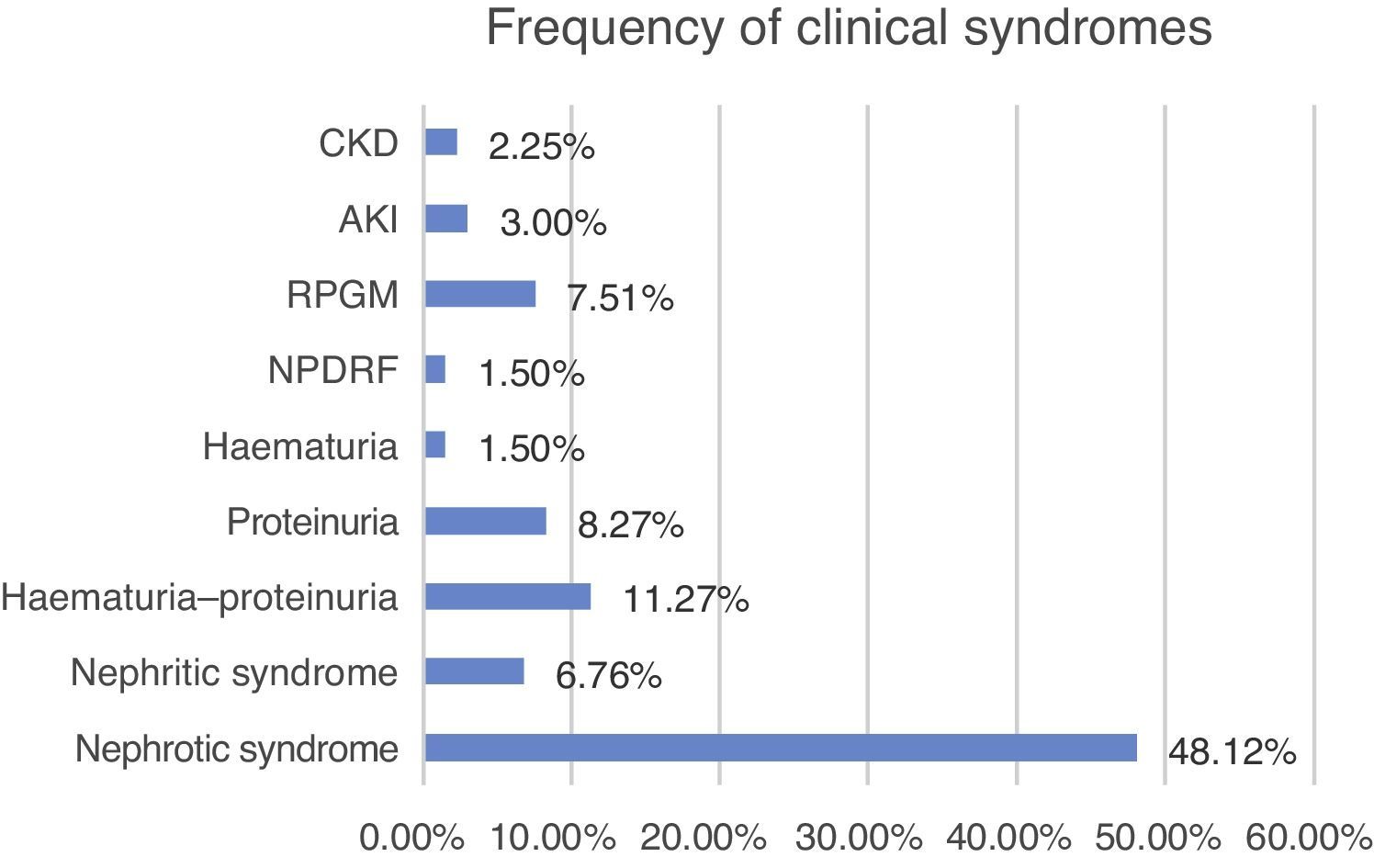
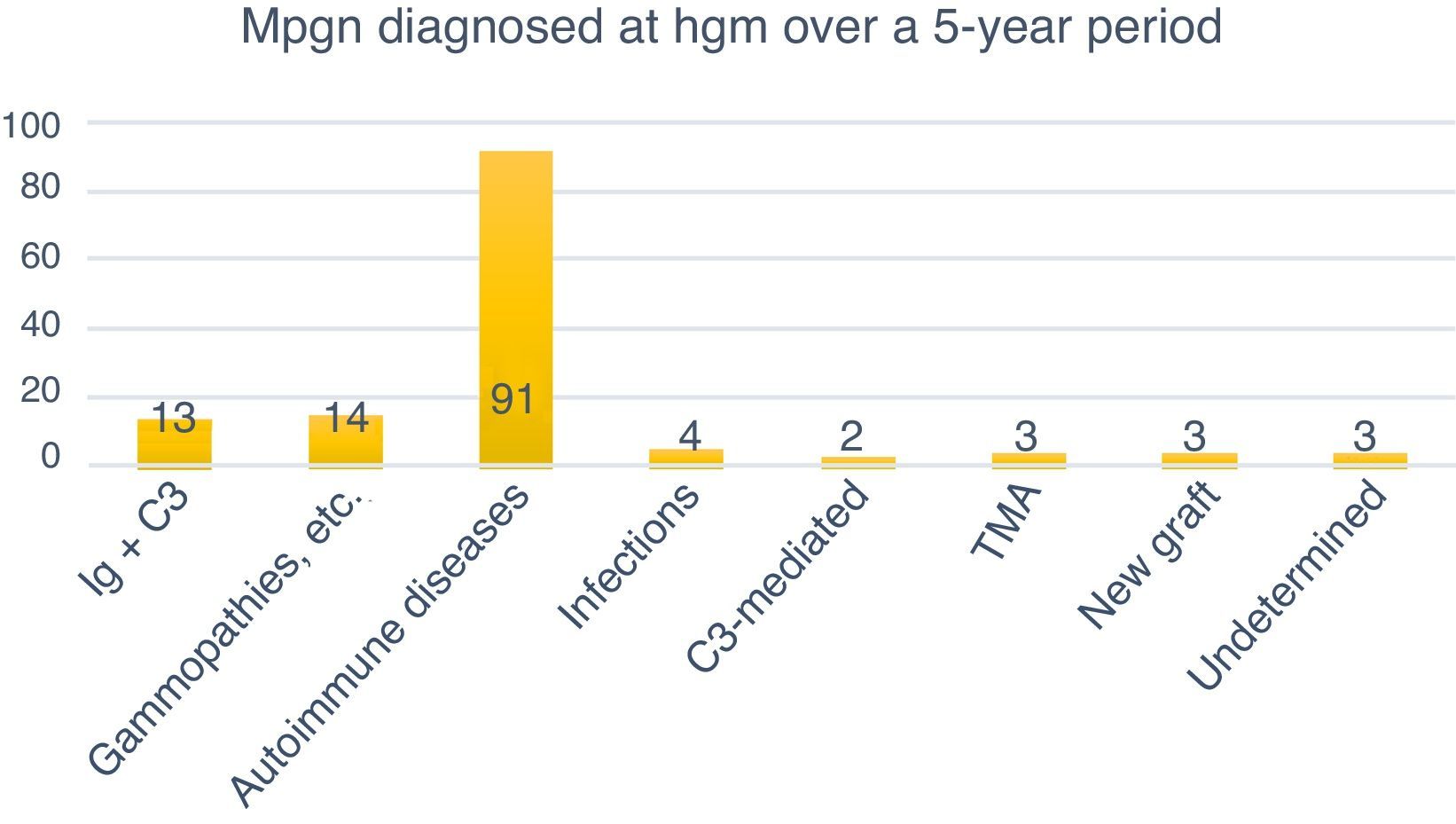
Results133 biopsies diagnosed as membranoproliferative glomerulonephritis were obtained. The average annual rate was 3.37%, while average age was 31.35±16 years. The disorder affected mostly women (60.15%, n=80). Nephrotic syndrome was the most common clinical presentation (48.12%, n=64), while autoimmune disease was the most common cause (77.77%, n=91).
ConclusionsDirect immunofluorescence is indispensable for classifying membranoproliferative glomerulonephritis.
La glomerulonefritis membranoproliferativa es un patrón de lesión glomerular con diferentes etiologías. Se clasifica por inmunofluorescencia directa en: glomerulonefritis membranoproliferativa mediada por complejos inmunes y glomerulonefritis membranoproliferativa mediada por complemento.
ObjetivoDeterminar la prevalencia de las glomerulonefritis membranoproliferativas mediante la biopsia (microscopía de luz, inmunofluorescencia directa y microscopía electrónica), ordenarlas de acuerdo a la más reciente clasificación, identificar las posibles etiologías y determinar algunas características epidemiológicas y clínicas.
Material y método. Se realizó un estudio transversal descriptivo donde se seleccionaron biopsias renales con patrón membranoproliferativo en 5 años en el Hospital General de México. Se obtuvieron edad, sexo, síndrome clínico y resultados de inmunofluorescencia y microscopía electrónica. Se excluyeron biopsias que carecieran de inmunofluorescencia y microscopía electrónica.
ResultadosSe obtuvieron 133 biopsias diagnosticadas como glomerulonefritis membranoproliferativa. La tasa anual promedio fue de 3.37%. La edad promedio fue de 31.35 ± 16 años. Las mujeres fueron las más afectadas (60.15%, n=80). El síndrome nefrótico fue la presentación clínica más frecuente (48.12%, n=64). La etiología más frecuente fueron las enfermedades autoinmunes (77.77%, n=91).
ConclusionesPara la clasificación de las glomerulonefritis membranoproliferativas es indispensable el estudio de inmunofluorescencia directa.
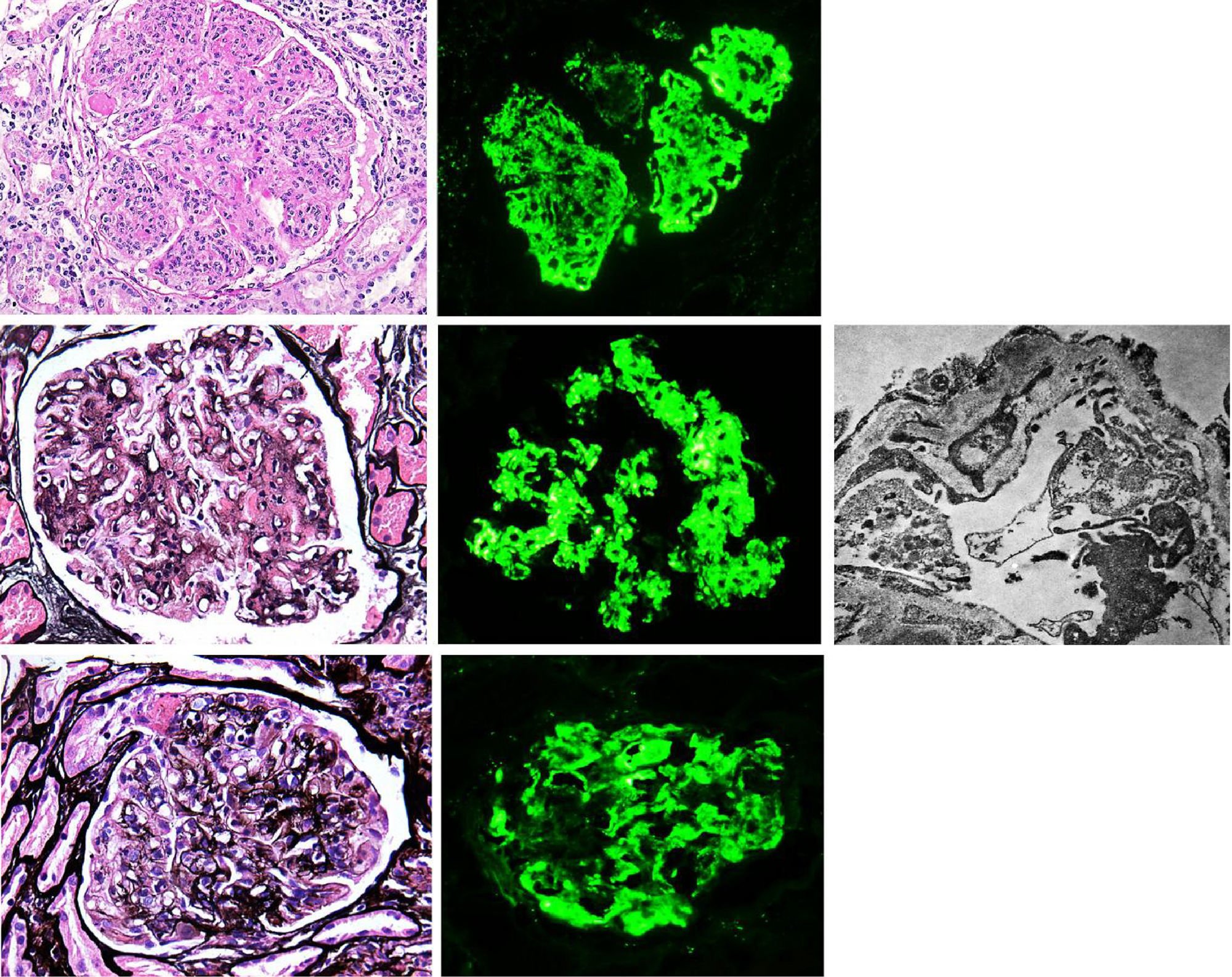
Membranoproliferative glomerulonephritis (MPGN), also called mesangiocapillary glomerulonephritis, is a pattern of glomerular lesion encompassing a variety of pathologically different glomerular diseases. In light microscopy (LM), MPGN is characterised by endothelial oedema, double contours due to duplication of glomerular basement membranes, cellular interposition and proliferative changes (Fig. 1). These changes occur as a result of the deposition of immunoglobulins, complement factors or both in the capillary wall and the mesangium.1 The causes for these types of glomerulonephritis vary according to geographic and environmental factors. In the US population, MPGN accounts for 7–10% of biopsied glomerular disease,2 whereas in developing countries (Asia, Africa and South America) it is present in 36.8%, with rural populations being the most affected. In Spain, it constitutes 4% of the population studied.3,1
 MPGN in Sjögren's syndrome. Homogeneous and diffuse lobular accentuation. (B) MPGN with C3-dominant deposits. EM in paraffin tissue block with subendothelial, membranous, mesangial and paramesangial electron-dense deposits. (C) Mixed cryoglobulinaemic glomerulonephritis. Membranoproliferative pattern with inflammatory cells, karyorrhexis and segmental fibrinoid necrosis. Direct immunofluorescence (DIF) was positive in the glomerular capillary lumen.")
(A) MPGN in Sjögren's syndrome. Homogeneous and diffuse lobular accentuation. (B) MPGN with C3-dominant deposits. EM in paraffin tissue block with subendothelial, membranous, mesangial and paramesangial electron-dense deposits. (C) Mixed cryoglobulinaemic glomerulonephritis. Membranoproliferative pattern with inflammatory cells, karyorrhexis and segmental fibrinoid necrosis. Direct immunofluorescence (DIF) was positive in the glomerular capillary lumen.
Traditionally MPGN was classified based on its histological characteristics and the electron microscopy (EM) findings, such as: MPGN type I, MPGN type II and MPGN type III; where type I is characterised by subendothelial electron-dense deposits, type II by electron-dense deposits in glomerular basement membranes and Bowman's capsule, and type III by the presence of subendothelial and subepithelial deposits.
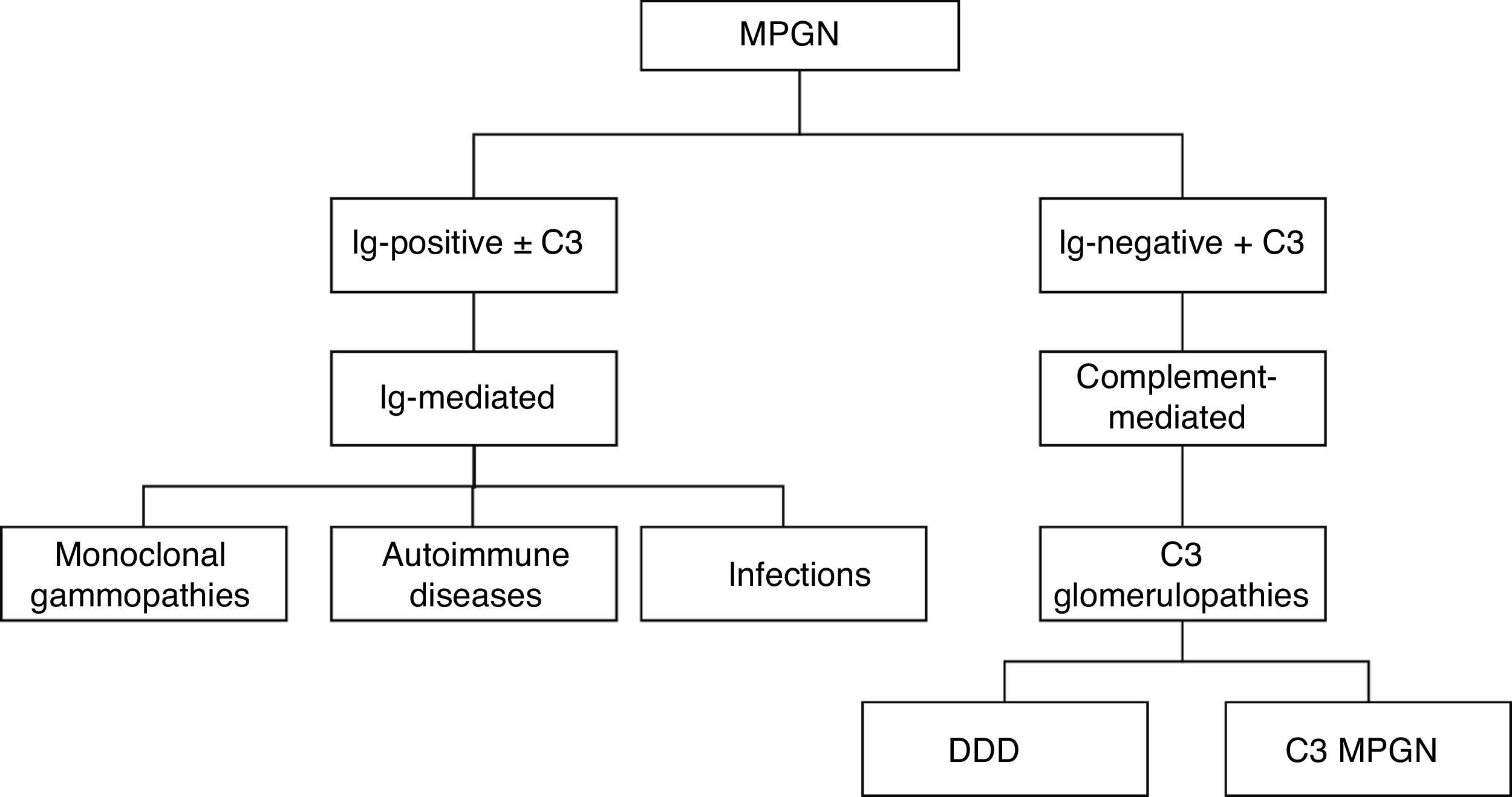
A new classification based on DIF results was formulated in 2015. This new classification has aetiological and therapeutic implications when considering two types: 1) immune complex-mediated MPGN with over-activation of complement, 2) complement-mediated MPGN. The former is divided into three subgroups—monoclonal gammopathies, autoimmune diseases and infectious diseases—while the latter corresponds to dense deposit disease (DDD) and C3 glomerulonephritis. MPGN symptoms with absence of deposits of immunoreactants suggest chronic thrombotic microangiopathy (TMA) (Fig. 2).

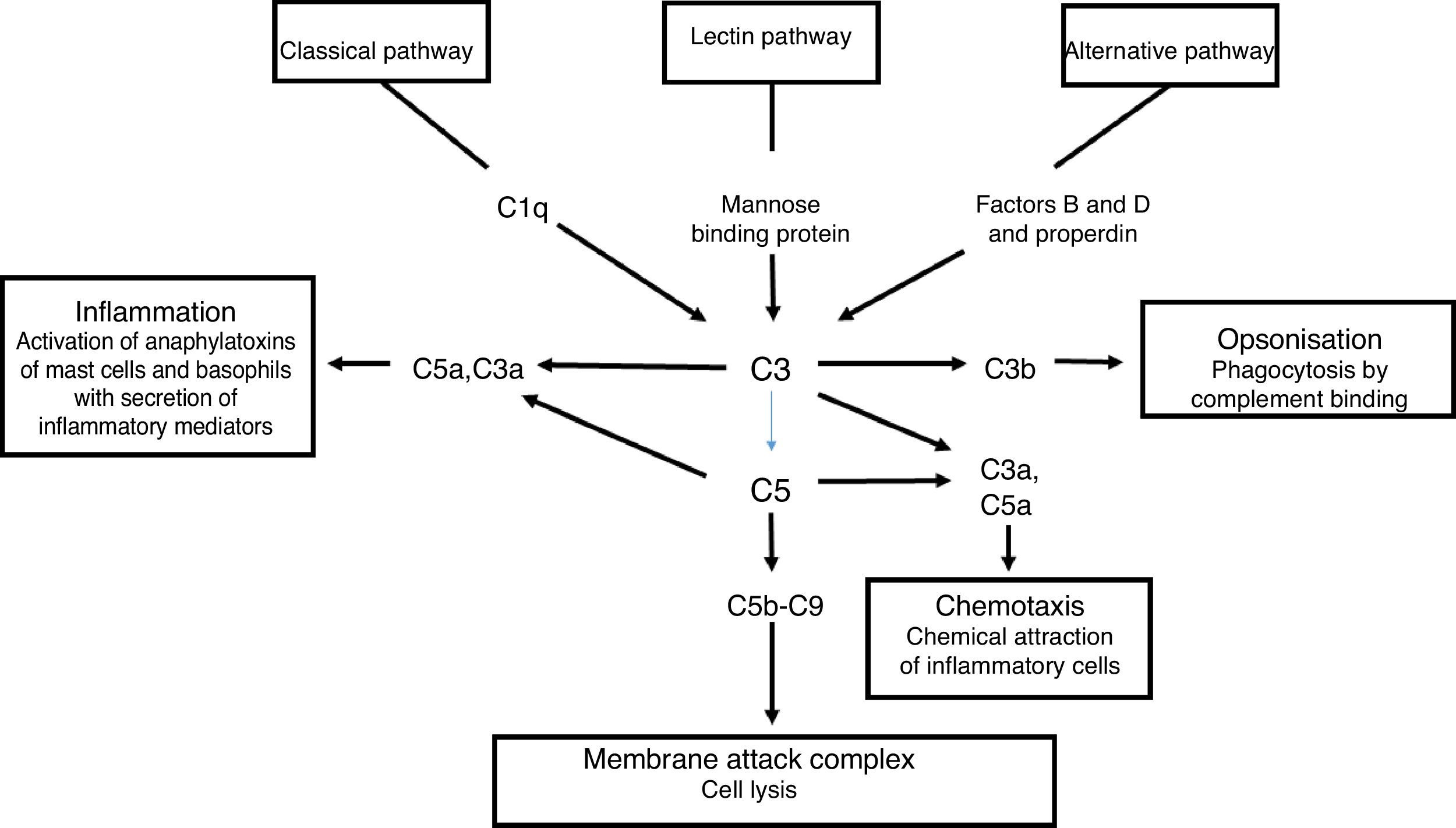
The complement system is a primary mediator of humoral immune response that allows the body to produce an inflammatory response, cause foreign cell lysis and increase phagocytosis. Uncontrolled activation of this system is prevented by inhibitor proteins and the instability of the activated complement proteins of the complete at each step of the process. There are three parallel but independent mechanisms for recognising microorganisms that result in the activation of the complement system: the classical, the alternative and the lectin pathways.4
Activation of the classical complement pathway is initiated by the interaction of C1q with an antigen-antibody complex. This interaction results in the formation of C4b2a which is the classical pathway C3b convertase. The alternate pathway uses C3 and factors B and D to form the C3b alternative pathway, Bb convertase. The lectin pathway begins once a mannose binding protein binds to mannose-containing molecules commonly present on the surface of bacteria and yeasts. Small amounts of C3b are constantly being formed in the circulation, which are inactivated by factors H and I. The binding of C3b to an antigen decreases its affinity for factor H and allows the formation of increasing amounts of alternative pathway convertase. The classical and alternative pathway convertases activate C3, forming C3a and C3b. C3b is an opsonin in its own right, and C3 convertase facilitates the activation of the terminal pathway and the formation of the C5b-9 membrane attack complex5 (Fig. 3).

Immune complex-associated MPGN is a result of the deposition of immune complexes in the glomerulus due to persistent antigenaemia. This leads to the formation of antigen-antibody complexes as a result of chronic infections, high levels of circulating immune complexes due to autoimmune diseases or monoclonal gammopathies.2
The term glomerulopathy with C3-dominant deposits is usually used to include C3 glomerulonephritis and DDD. It may be difficult to distinguish them in LM or DIF, but EM shows mesangial and/or subendothelial, intramembranous and subepithelial deposits in C3 glomerulonephritis, and osmiophilic dense deposits along the glomerular basement membrane and in the mesangium in DDD.4 DDD is characterised by the deposition of dense material inside the glomerular basement membrane, Bowman's capsule and the tubules. These deposits do not contain immunoglobulins, but appear to activate the alternative complement pathway. The presence of hypocomplementaemia in DDD reflects the activation of the disorder. Three mechanisms result in the uncontrolled activation of C3 convertase: 1) development of the antibody C3 nephritic factor (C3NeF); 2) absence of circulating factor H; and 3) presence of a circulating factor H inhibitor. The most common mechanism is the presence of the C3NeF autoantibody, which protects C3 convertase (C3bBb) from dissociation by factor H, thereby prolonging its half-life ten-fold. It does this in two ways: by binding to either C3bBb or IgG-c3b-C3bBb of the assembled convertase. The stabilisation of this complex results in the perpetual failure of C3.7
MPGN without immune complexes or complement is a pattern that can also be seen in TMA. There are several causes that can lead to this pattern, such as systemic infections, neoplasms, haemolytic–uraemic syndrome/thrombotic thrombocytopenic purpura (resolved), antiphospholipid syndrome, radiation nephropathy (related to bone marrow transplantation) and transplant glomerulopathy, among others. Regarding transplant glomerulopathy, the absence of C3 and immunoglobulins in DIF can help to distinguish it from recurrent MPGN,6 while the presence of peritubular capillaritis and C4d positivity in peritubular capillaries distinguish it from chronic TMA.
The recurrence of MPGN has been reported after renal transplantation in previous studies. Based on pathogenesis, the rates reported vary, ranging from 27% to 65%.8 Post-transplant recurrence has been associated with low levels of complement, severe proteinuria, related living donor, half-life in the original biopsy and presence of monoclonal gammopathy.9
This study aimed at determining the prevalence of MPGN by studying LM, DIF and EM biopsies at Hospital General de México “Dr. Eduardo Liceaga” (HGM), categorising it according to the most recent classification and identifying the possible causes, thus determining certain epidemiological and clinical characteristics.
MethodsA descriptive, cross-sectional study was carried out using native kidneys biopsies and grafts with histopathological diagnosis of membranoproliferative glomerulonephritis (or mesangiocapillary glomerulonephritis) performed between June 2011 and June 2016 at Hospital General de México. Age, gender, clinical syndrome and DIF and EM results were obtained. Biopsies lacking DIF or EM results were excluded. Data were analysed using descriptive statistics. Quantitative endpoints were presented as mean, standard deviation, and minimum and maximum values. Qualitative endpoints were presented as frequencies and percentages.
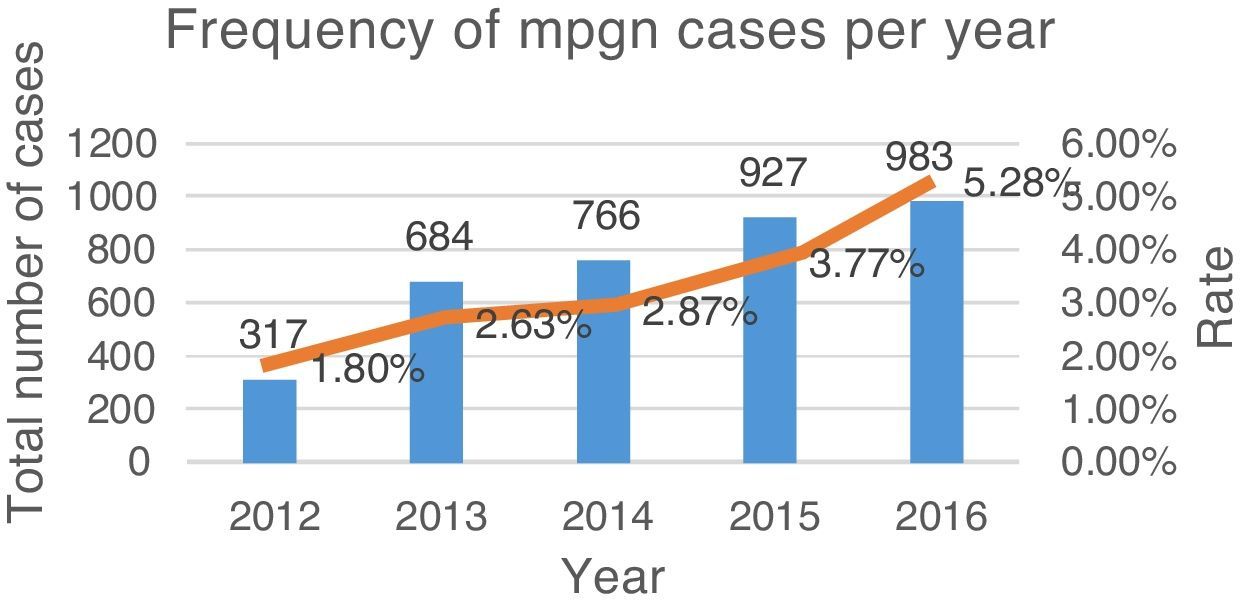
ResultsFrom June 2011 to June 2016, a total of 3677 renal biopsies were received, of which 133 (3.6%) were diagnosed as MPGN. The average annual rate was 3.37% of the total biopsies studied (Fig. 4). Of these, 124 (93.23%) corresponded to native kidney biopsies and four (3.0%) to renal grafts, while no data were obtained for five (3.75%). The most common type of biopsy was percutaneous puncture, corresponding to 93.98% (n=125) of the total biopsies, followed by open-wedge biopsy (3.0%, n=4), case reviews (2.25%, n=3) and one (0.75%) nephrectomy. Because all cases (n=133) lacked complete clinical information, the study of the clinical endpoints (creatinine, haemoglobin, viral profiles, immunological profiles, etc.) could not be performed. 93.98% (n=125) included DIF, the cases that did not include DIF correspond to cases sent for review (2.25%, n=3), while the rest (3.75%, n=5) had undergone formalin fixation. EM was only performed in 27 (20.3%) cases; the study could not be performed in the rest (79.69%, n=106) because the material was inadequate.

Of the 133 patient records, only 131 (98.49%) include age. The average age of these patients was 31.35±16 years, with a minimum age of 1 year and a maximum age of 74 years. The disorder affected mostly women (60.15%, n=80) while the corresponding biopsies for men constituted 39.84% (n=53) of the total population.
Clinical characteristics at the time of biopsyThe biopsied patients were grouped as follow, with nephrotic syndrome being the most common clinical presentation (48.12%, n=64), followed by haematuria–proteinuria (11.27%, n=15), proteinuria (8.27%, n=11) and rapidly progressive glomerulonephritis (7.5%, n=10) (Fig. 5).
Classification of MPGN according to DIF
The biopsies were organised according to the new classification using DIF in two groups: those mediated by immune complexes (93.98%, n=125) and those mediated by complement (1.5%, n=2). Autoimmune diseases were the most common cause for immune complex-mediated glomerulonephritis (77.77%, n=91). Three (2.25%) cases corresponded to TMA, while it was not possible to determine the cause in another three (2.25%) due to chronicity. Of the 133 cases, four (3%) were renal grafts, two of which (1.5%) corresponded to de novo MPGN with onset in the graft at an average of 7.6 years after transplantation. In one (0.75%) case, disease recurrence was confirmed at 8.3 years after transplantation, while another (0.75%) case corresponded to glomerulonephritis with C3-dominant deposits (Fig. 6).
Discussion
In our study, MPGN had a prevalence of 3.37%, which is lower than that reported in the North American, Asian and African literature.2,3 Immune complex-mediated MPGN was more common with 125 (93.98%) cases, which correlates with what was reported in the literature.10,11 In this group, systemic lupus erythematosus was the most common autoimmune disease with 75 (60%) cases, of which 46 (61.33%) were women aged 1–64 years, with an average age of 26 years±13.67 and a prevalence at childbearing age, as reported in the literature.11 It is noteworthy that these autoimmune causes were the most common in our study because the child population in our hospital is smaller. Three (2.4%) cases were diagnosed with Sjögren's syndrome, which was later corroborated with laboratory tests and clinical studies. Mixed cryoglobulinaemia was the second most common disease in this group with 6.4% (n=8) of the cases studied, with one (0.8%) of these being reactive for hepatitis C virus. In the literature it was reported that most cases of mixed cryoglobulinaemia are found in patients with connective tissue diseases, infectious diseases, lymphoproliferative disorders, hepatobiliary diseases or immune complex-mediated glomerular diseases,12 which is consistent with our findings.
We found two cases with fibrillary glomerulonephritis, which constituted 1.6% of the total cases assessed, corresponding to the literature reports. These cases were characterised by IgG, IgA, C3c, kappa and lambda positivity which, according to published series, would be positive for IgG and C3, 60% for IgM and 30% for IgA. Both kappa and lambda chains are positive in most patients.12 Of the total cases assessed (n=133), two of four grafts had a prior history of MPGN which reoccurred, unlike the literature, which reports a 67% recurrence versus 100% in our study. This can be explained by the small number of cases (two) included in our study, compared with the 18 cases reported by Braun et al.13
In addition, in the complement-mediated glomerulonephritis group, two cases (1.5%) were observed that corresponded to C3-dominant deposits. According to literature reports, this disease occurs in both genders at different ages.4 Our two cases included a 35-year-old male patient and a 12-year-old female patient at the time of diagnosis.
Although the number of cases was small, it was possible to observe that our results correspond to what is described in the world literature. Unlike Asian, African and South American countries where prevalence is higher, we believe the lower prevalence observed in our study may be due to the fact that in Mexico glomerulopathies where the cause is clear, either by DIF or EM, are separated, and we diagnose according to histological pattern only those for which we do not have sufficient clinical and laboratory data to make a definitive diagnosis. In developed countries such as the United States, where renal biopsy is a confirmatory diagnostic tool rather than a means to guide the clinician to continue with the study protocol, lower prevalence is reflected in the lower number of cases reported. Moreover, the lack of laboratory data was an impediment to analysing clinical endpoints and correlating with diagnosed cases. However, the information obtained in this study was valuable as an overview of how MPGN is found in the studied population.
In view of the above, we conclude that:
- –
Direct immunofluorescence is indispensable for classifying MPGN, followed by electron microscopy, which is an important tool. These mainly allow for distinguishing cases that involve C3-dominant deposits.
- –
Clinical information is indispensable for making a more specific diagnosis.
The authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
FundingDepartment and own resources.
Conflict of interestThe authors declare that they have no conflicts of interest.
We would like to thank the Department of Pathology at Hospital General de México Dr. Eduardo Liceaga for providing the facilities to perform this study.