


Sertoli–Leydig cell tumours (SLCT) are rare tumours, accounting for less than 0.5% among the ovarian tumours, and occur most frequently in the second and third decades. The majority of them are unilateral and diagnosed at stage I. We report the case of an 11-year-old girl with an abdominal mass and virilisation; left salpingo-oophorectomy was performed and an ovarian SLCT of intermediate differentiation was diagnosed. Twenty nine months later, a new abdominal mass was discovered; a salpingo-oophorectomy was performed and a second SLCT, poorly differentiated, was diagnosed. These tumours represent a rare entity in adolescents and girls, and the bilateral and metachronic presentation makes it exceptional. Despite its low incidence, a high rate of suspicion should be given in the diagnosis of these tumours in very young patients with or without signs of virilisation, and strict surveillance should be maintained in the search for a new related tumour.
Los tumores de células de Sertoli-Leydig (TCSL) son neoplasias inusuales, con una frecuencia de menos del 0.5% de todos los tumores ováricos y se suelen presentar entre la segunda y tercera décadas. La mayoría son unilaterales y se diagnostican en estadio I. Reportamos el caso de una paciente de 11 años con masa abdominal y características de virilización, se le realiza salpingooforectomía izquierda diagnosticándosele un TCSL ovárico de diferenciación intermedia. A los 29 meses siguientes, presenta otra masa abdominal; se realiza salpingooforectomía y es diagnosticada como un segundo TCSL de pobre diferenciación. Estos tumores representan una rara entidad en niñas y adolescentes, y la presentación ovárica bilateral y metacrónica lo hace excepcional. A pesar de su baja incidencia, se debe tener una alta sospecha para su diagnóstico en pacientes muy jóvenes con o sin signos de virilización, además se debe mantener una estricta vigilancia por la posibilidad de un nuevo evento tumoral relacionado.
Sertoli–Leydig cell tumours (SLCTs) are rare neoplasms belonging to the sex cord–stromal tumour group. They account for less than 0.5% of ovarian tumours in general and mainly occur in young adult women in their 20s.1–3 These tumours, which account for just 4% of all ovarian tumours in women under 20 years of age, are very rarely diagnosed before menarche.4 Their clinical signs may be due to abnormal production of hormones, especially androgens, or the presence of a mass in the pelvic/abdominal region.5 Nearly all are unilateral; bilaterality and metachronism are extremely rare characteristics, being observed in 1.5% of cases.6 Less differentiated histological subtypes are most commonly observed in young patients, and the retiform histological pattern is most commonly reported at an average age of 16 years, compared to 26 years for the diagnosis of SLCTs in general.7 Treating young patients is a challenge. It is essential to achieve complete healing and preserve fertility while avoiding mutilating procedures.8 We report the rare case of a bilateral, metachronous ovarian SLCT in an 11-year-old patient, who developed the tumour on her right ovary 29 months after she underwent primary surgery on her left ovary. We also review the existing literature, including the literature on tumour characteristics, prognostic factors and treatment.
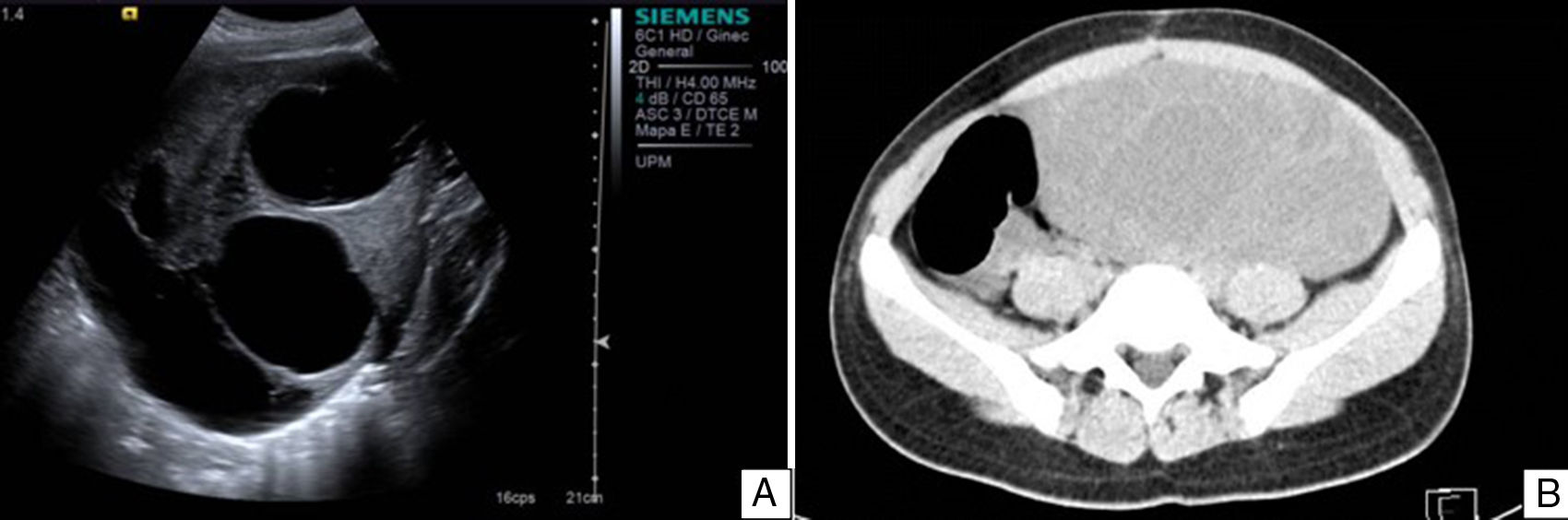
Case reportAn 11-year-old premenarchal female patient visited the Paediatrics Department with clinical characteristics of virilisation (deep voice, hirsutism of the lower limbs and moderate clitoromegaly) for the last 7 months, associated with a palpable mass on the left iliac fossa. A pelvic ultrasound revealed a lesion on the left uterine adnexa of 102×88×62mm, initially interpreted as “cystadenocarcinoma”, despite her age (see Fig. 1). The dimensions of the right ovary were 30×22×15mm. The uterus was normal. A hormone panel revealed that the patient had a slightly increased level of testosterone, i.e. 81ng/dl (acceptable values below 75ng/dl). Her levels of progesterone, oestradiol, luteinising hormone and follicle stimulating hormone were within normal limits. Her levels of other tumour markers (α-fetoprotein, lactate dehydrogenase, beta fraction of human chorionic gonadotropin [hCG], Ca-125 and carcinoembryonic antigen) were within normal limits as well. An exploratory laparotomy showed an ovary of 11cm at its greatest diameter occupying the left iliac fossa. The ovary was mobile, with no evidence of adhesions. A unilateral salpingo-oophorectomy was performed. The uterus and right adnexa showed no abnormalities. The specimen was sent to the Pathology Department.

A. Pelvic ultrasound of the left tumour. Well delimited solid–cystic lesion of 102×88×62mm with limited vascularity. No other abnormalities. B. Abdominal and pelvic CT scan of right ovarian lesion with characteristics similar to those found on ultrasound, displacing intestinal loops and partially compressing the right ureter.
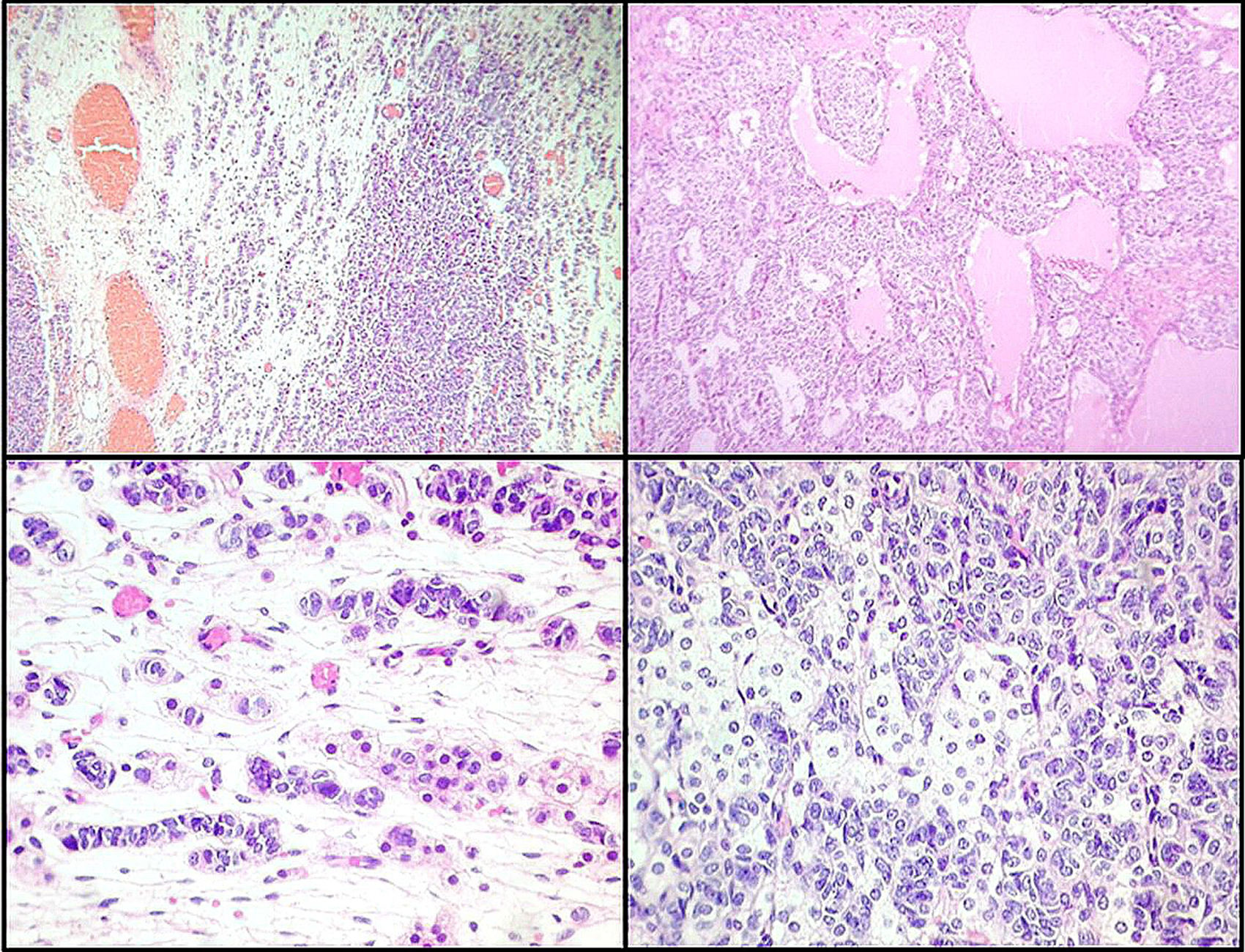
Macroscopically, the ovarian mass measured 11×7.5×7cm, with a smooth, intact, bright capsule. The cut surface was solid/cystic, with some yellow areas and other areas with a myxoid appearance. Microscopically, a lesion was identified that consisted of two cell groups, largely consistent with immature Sertoli cells arranged in lobes and trabeculae with more limited intermixed groups of Leydig cells, with abundant eosinophilic or clear cytoplasm. It was diagnosed as a stage T1a Sertoli–Leydig cell tumour of intermediate differentiation, without heterologous elements, with an intact capsule and without implants (see Fig. 2). The patient was not considered a candidate for chemotherapy.
. A. Solid lobular areas are observed, alternating with hypocellular areas of lax stroma with trabecular and linear formations. B. Structures with a tubular appearance and cystic areas containing homogeneous eosinophilic material are identified between solid areas. C and D. Sertoli cells arranged in a trabecular pattern. Areas of Leydig cells with a central nucleus, a single nucleolus and eosinophilic to clear cytoplasm are observed.")
Left ovarian tumour (first tumour). A. Solid lobular areas are observed, alternating with hypocellular areas of lax stroma with trabecular and linear formations. B. Structures with a tubular appearance and cystic areas containing homogeneous eosinophilic material are identified between solid areas. C and D. Sertoli cells arranged in a trabecular pattern. Areas of Leydig cells with a central nucleus, a single nucleolus and eosinophilic to clear cytoplasm are observed.
During her 19 months of follow-up she showed normal hormone levels, and during the year of her surgical procedure she reached menarche. After 10 months with no record, the patient was readmitted with a pelvic mass (29 months after her first surgery); this time, she presented no clinical characteristics of virilisation or menstrual irregularities. Ultrasound and an axial CT scan demonstrated a multicystic, heterogeneous right ovarian tumour of 201×184×164mm (see Fig. 1). The patient was found to have increased serum levels of Ca-125 and α-fetoprotein and levels of testosterone, progesterone, oestradiol, luteinising hormone, follicle-stimulating hormone, α-fetoprotein, lactate dehydrogenase, beta fraction of HCG, Ca-125 and carcinoembryonic antigen within normal limits. An exploratory laparotomy showed an ovary of 23.5cm at its greatest diameter. The ovary was mobile, with no evidence of adhesions. It occupied the entire right iliac fossa and displaced the intestinal loops, and was found to be in contact with the sigmoid colon and in close proximity to the abdominal aorta. A right salpingo-oophorectomy and omentectomy were performed, and the specimens were sent to the Pathology Department.
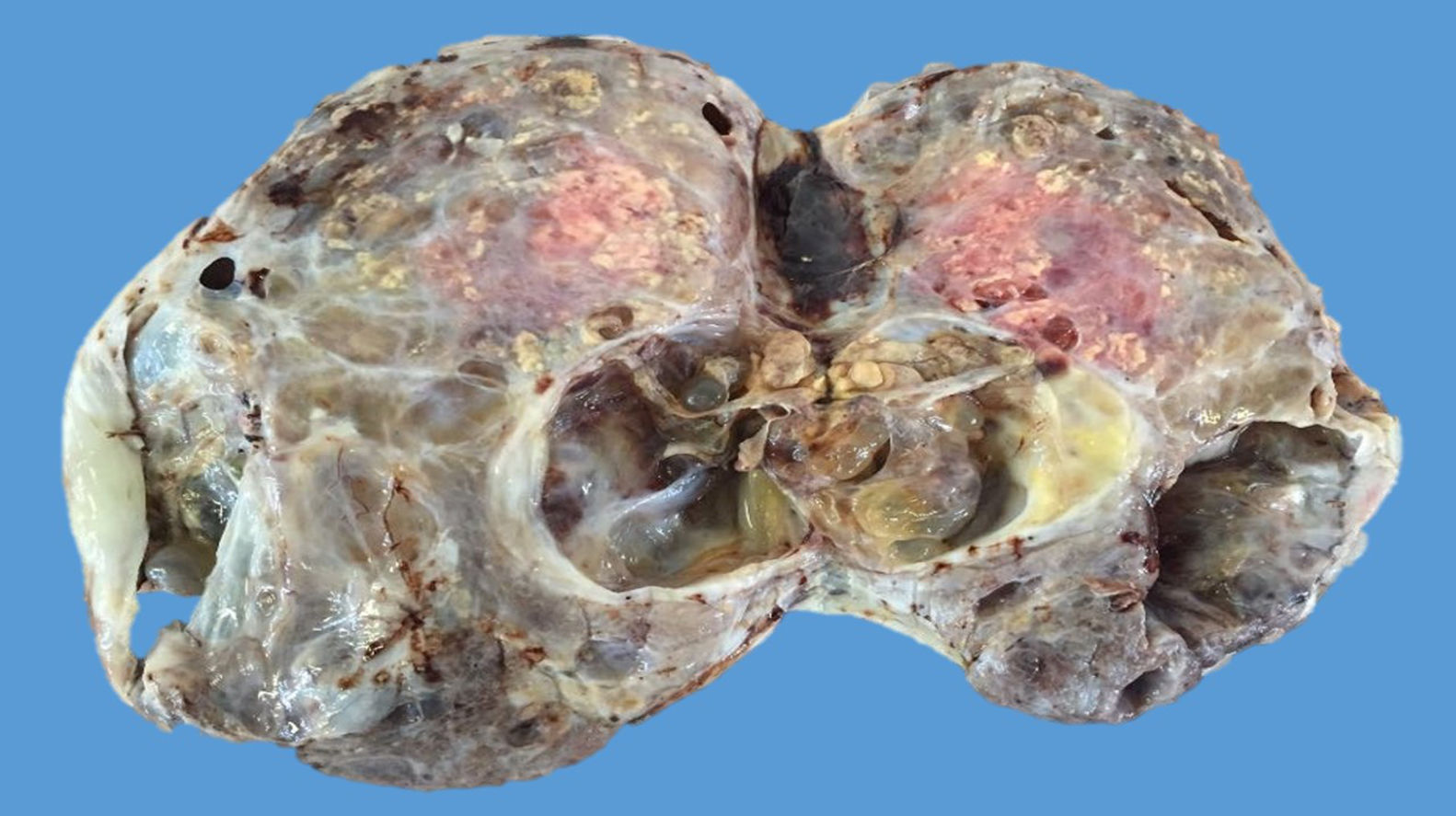
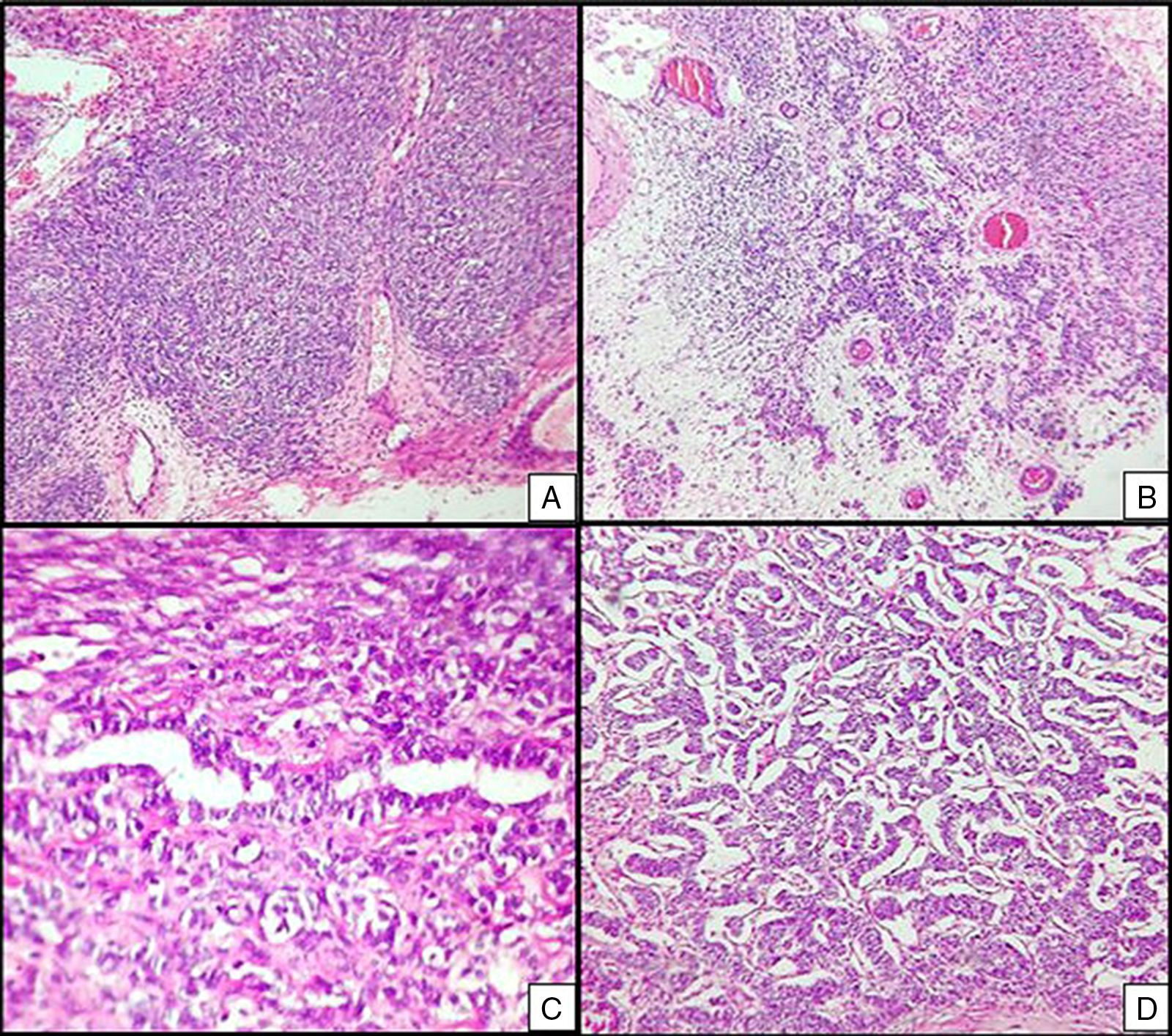
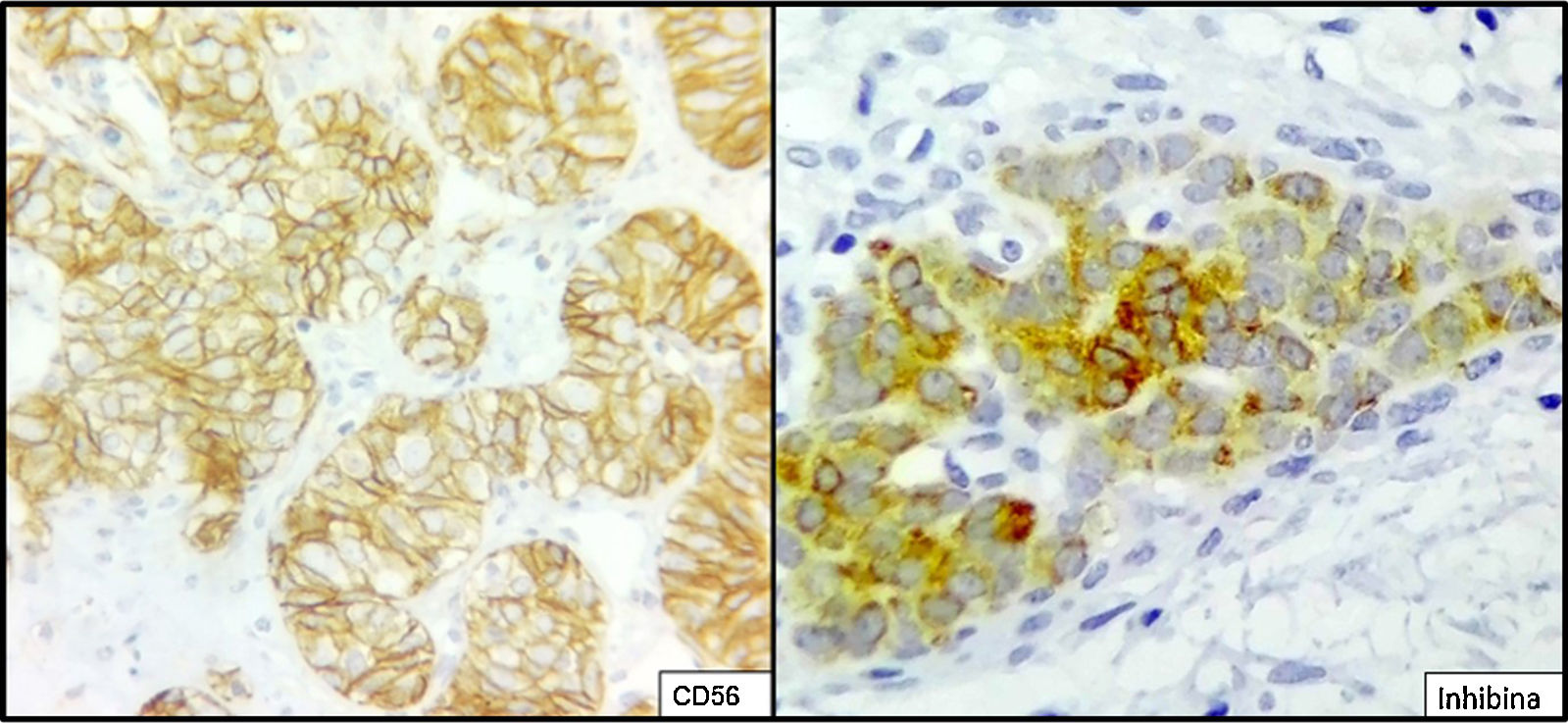
The ovarian mass weighed 500g and measured 23.5×18.8×7.5cm. It had a smooth, bright, intact capsule. The cut surface was heterogeneous, predominantly (70%) occupied by solid brown areas, with cystic areas (see Fig. 3). The omentum measured 12×5cm and had no macroscopic abnormalities. Microscopically, the tumour had a solid sarcomatoid pattern of Sertoli cells with formation of focal tubules and scarce Leydig cells (see Fig. 4). The Sertoli cells showed positivity for inhibin and CD-56 (see Fig. 5). The mass was diagnosed as a poorly differentiated stage-T1a Sertoli–Leydig cell tumour with an intact capsule and without implants. No further treatment was administered. After 18 months, the patient was asymptomatic, with no imaging or laboratory abnormalities. She remains under surveillance.


Right ovarian tumour. Microphotographs are observed with multiple patterns in the same tumour: A. Solid poorly differentiated sarcomatoid areas consisting of short fusiform cells with randomly organised hyperchromatic ovoid nuclei. B. Areas with a more classic appearance of a Sertoli–Leydig cell tumour, with solid areas and areas of Sertoli cell trabeculae interconnected with each other alternating with areas of dispersed cell groups on a lax background. C. Tubular areas similar to those observed on the left ovarian tumour, arranged in a dispersed fashion, with some between the sarcomatoid areas and D. areas with a trabecular–gyriform pattern.

Ovarian sex cord–stromal tumours are uncommon neoplasms of the upper female genital tract that account for 0.5% of all ovarian neoplasms.3 They include SLCTs, which have a frequency of 0.2–0.5%.3,9 They have been reported in patients from 6 months to 84 years of age, but are most commonly diagnosed in young women 25–28 years of age, except the retiform variant, which has a worse prognosis and is diagnosed at 16 years of age on average.1,5,9 Less than 10% are diagnosed in premenarchal and postmenopausal patients.9 They are typically unilateral and confined to the ovary. Just 1.5% are bilateral.1,10
These tumours should always be suspected in young women with a pelvic mass under study and/or signs and symptoms of virilisation.3 However, it should be borne in mind that 20% of cases are too small to be detected by imaging techniques.1 Signs of excess androgens, with elevated serum testosterone levels, are identified in up to 50% to 60% of patients.9 Initially, there are aspects of defeminisation such as amenorrhoea; later, there are aspects of virilisation such as breast atrophy, hirsutism, deepening voice, laryngeal protuberance and enlargement of the clitoris. The association between virilisation and the degree of histological differentiation of these tumours is debated.1 More rarely, symptoms are due to excess production of oestrogens, which may result in early puberty, abnormal uterine bleeding, menstrual irregularities and endometrial hyperplasia or adenocarcinoma.11 Our patient had some classic characteristics of virilisation associated with an abdominal mass during her first tumour event. By contrast, her second tumour event was only characterised by a mass even larger than the first one.
Preoperative diagnosis is difficult; however, it may be suspected in cases of progressive endocrine symptoms and a pelvic mass. Increased plasma testosterone levels >200ng/dl are nearly always related to an ovarian neoplasm or androgen-secreting adrenal neoplasm.1 Levels of 17-ketosteroids in urine are usually normal or slightly elevated in these patients, as opposed to those with virilising adrenal tumours.12 Combined with ultrasound, which is the preferred imaging modality for initial evaluation of adnexal masses, a CT scan, magnetic resonance imaging (MRI) or positron emission tomography (PET) may offer an examination with better visualisation of the ovaries and other abdominal organs.5,12
SLCTs are generally heterogeneous. They may be solid, cystic or mixed. The latter macroscopic appearance is found in up to 60% of cases. The average major axis tends to be 13.5cm; some may be impossible to detect by ultrasound and others may measure up to 50cm.6 They are tumours that develop from the non-germ cell component of the ovary, in which the elements recapitulate to a greater or lesser extent the cells of the stroma–sex cords in variable degrees of differentiation: primitive gonadal stroma, epithelial cells of the rete ovarii and/or heterologous elements.2,10 They are divided into five histological types: 1—well differentiated, which are the least common and generally have benign biological behaviour; 2—of intermediate differentiation, which are the most common; 3—poorly differentiated, 4—with heterologous elements and 5—with retiform elements.13 It has been observed that with a lower grade of differentiation, the tubular architecture with Sertoli cells is less prominent and the nuclear atypia, the number of mitoses and the areas of necrosis and bleeding are more common. Heterologous elements are present in up to 3–10% of these tumours, being observed in those with intermediate or poor differentiation, with tissue elements not corresponding to the sex cords, most commonly mucinous epithelial3,4 in addition to foci of cartilage and skeletal muscle. The retiform variant is observed in 10–15% of cases and has unique microscopic and clinical features: it features a papillary–tubular pattern, is common in young patients, may produce α-fetoprotein and carries a worse prognosis.5,10 This component should be observed in more than 90% of the tumour for it to be classified as retiform; otherwise, it is diagnosed as part of an intermediate-grade or poorly differentiated tumour.4
Due to their multiple histological patterns, SLCTs lend themselves to numerous differential diagnoses: endometrial carcinoma, carcinoid tumours, teratoma and other sex cord–stromal neoplasms (e.g. granulosa cell tumours). Immunostaining is useful in some cases, particularly poorly differentiated variants and variants with atypical patterns. Sertoli cells are positive for calretinin, inhibin, CD-56, cytokeratins, CD99, WT1, SF-1 and CD-10.5,6,14,15 Leydig cells are positive for vimentin and melan-A.16 Endometrioid carcinoma, for example, is negative for antibodies against inhibin and calretinin, and is positive for epithelial membrane antigen.15 Positivity for CD-56 is shared with carcinoid tumours, whose trabecular architecture may lend itself to confusion. However, SLCTs’ negativity for other neuroendocrine markers, such as synaptophysin and chromogranin and positivity for those already indicated helps to differentiate them.14 Within the ovarian sex cord–stromal tumour group, only SLCTs in their Leydig cell component and steroid cell tumours express melan-A.14
The standard management guideline for ovarian SLCTs remains uncertain. The recommended treatments vary based on age, tumour stage and tumour differentiation. The initial treatment method is a surgical approach.17 The management suggested for young patients with early stages (T1a/b) wishing to preserve their fertility is unilateral salpingo-oophorectomy with or without examination of the contralateral ovary. Some authors have promoted minimal resections, especially in small, well encapsulated lesions, followed by oophorectomy or salpingo-oophorectomy depending on histopathological results. However, in general, they are not favoured to prevent incomplete resections and tumour rupture.8 For those with stage T1 with tumours of intermediate or poor differentiation, or with capsule rupture, standard staging (omentectomy, appendectomy and pelvic lymphadenectomy) should also be performed. Total hysterectomy and bilateral salpingo-oophorectomy or cytoreductive surgery should be recommended for women not wishing to preserve their fertility or with advanced SLCTs.1,3 Platinum-based postoperative chemotherapy is indicated in patients with poorly differentiated SLCTs and SLCTs with heterologous elements. However, there is very little information on an optimal adjuvant regimen.3
Patients must remain under surveillance with serum testosterone levels measured every 3 months for one year, every four months for the second year, every six months for the third year and every year from the fourth year for the rest of their lives. During follow-up, pelvic and abdominal ultrasounds should also be performed.17
Unlike other more common tumours of the stroma and sex cords, which recur late, relapse with SLCTs is early; 2/3 of cases recur within a year of the initial diagnosis, and just 7% recur after 5 years, with nodules in the retroperitoneum and abdominal cavity.3 These events are more strongly linked to preoperative and intraoperative capsule rupture.10 All patients, especially those with bilateral tumours, should also be screened for thyroid disease during follow-up, due to the link between SLCTs and DICER1 gene mutations.18
According to recently reported clinical and pathological data, mean age at diagnosis of tumours associated with mutations of this gene is 16 years (range of 10–28 years), tumour dimensions range from 35mm to 300mm and tumours are most commonly classified as SLCTs of intermediate differentiation and present heterologous elements more commonly than sporadic SLCTs.19 These mutations are also associated with the appearance of pleuropulmonary blastomas, familial multinodular goitre and, less commonly, renal and central nervous system tumours. Therefore, genetic counselling should be recommended, as a mutation in this gene has implications for follow-up.10 Our patient was not evaluated to identify this mutation, although her age and diagnosis of bilateral SLCT supported the suspicion that she was a carrier of the mutation.
The most important prognostic factors in these tumours are stage and the degree of differentiation. Tumours in an advanced stage have a worse prognosis with a mortality rate of 100%. According to Young et al. in a review of 207 cases, 11% of tumours with intermediate differentiation, 59% of tumours with poor differentiation and 19% of tumours with heterologous elements are malignant, whereas all well differentiated tumours are benign.2,20 Mesenchymal elements, such as smooth muscle, and epithelial elements, such as foci with a retiform pattern, carry a worse prognosis.21 In addition, production of α-fetoprotein, which has been associated with the retiform pattern, should be borne in mind as a prognostic factor.10
Two cases in the literature have reported the asynchronous appearance of a new SLCT on the contralateral gonad. In one case, the pathology was consistent with a poorly differentiated tumour, which recurred both on the opposite ovary and on the pelvis adjacent to the rectum and urinary bladder.22 In the other case, a 14-year-old girl with an SLCT of intermediate differentiation had a tumour, also of intermediate differentiation, on the contralateral gonad 40 months after her initial diagnosis.23 Other cases of synchronous SLCTs have previously been reported in patients 1724 to 1825 years of age.
ConclusionWe have presented a case of SLCT that was exceptionally rare for two reasons. First, it occurred in a preadolescent, premenarchal patient. Second, it was bilateral and metachronous: the first tumour presented with intermediate differentiation and the second tumour subsequently presented with poor differentiation. Due to its rarity, the diagnosis was not clinically suspected, despite its classic presentation, and could only be made by means of a histopathological study in both cases. No mutation that increased the risk of developing any new tumours was found. The patient had only been treated surgically, by means of a bilateral salpingo-oophorectomy and omentectomy with no neoadjuvant therapy, and required strict follow-up due to her prognostic factors.
Ethical disclosureProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThere are no conflicts of interest in the preparation or the publication of this document.