





Las enfermedades neuromusculares son un grupo heterogéneo de patologías consideradas raras, que en su conjunto afectan a un grupo importante de la población.
Las manifestaciones clínicas son variadas según la enfermedad con compromiso de distintos sistemas, muchas veces graves y generadoras de discapacidad, que pueden llevar a la muerte. Los avances terapéuticos en los últimos años han cambiado la evolución natural de algunas de estas enfermedades mejorando la expectativa de vida. A esto se suma el abordaje por equipo multidisciplinario, con intervenciones terapéuticas basadas en consensos internacionales, identificando precozmente y previniendo complicaciones potencialmente modificables. Esto representa un desafío para los equipos de salud, especialmente para el equipo de rehabilitación para mejorar la calidad de vida del enfermo y su familia promoviendo la inclusión escolar, laboral y social. En este artículo se realiza una revisión sobre aspectos respiratorios, nutricionales, cardiológicos y del abordaje de rehabilitación de estas enfermedades.
Neuromuscular diseases are a relatively rare and heterogeneous group of disorders that, as a whole, affect a significant part of the population. The spectrum of signs and symptoms vary with involvement of different organs leading to disability and sometimes death. Recent therapeutic developments changed the course of the disease and prolonged patient's survival. In addition, multidisciplinary groups participate in the treatment of these patients, applying international approved methodologies, for the early identification and prevention of potential complications. Responding to these advances represent a challenge for therapy-intervention-groups, and in particular rehabilitation teams. These aims should be met to improve the lifestyle of patients and relatives by promoting effective education, enhance work possibilities and social integration. In this article we review respiratory, nutritional, cardiology and rehabilitation strategies.
Las enfermedades neuromusculares (ENM) son un grupo heterogéneo de patologías que se caracterizan por afectar el asta anterior de la médula espinal, el nervio periférico, la unión neuromuscular o el músculo. En su mayoría son de origen genético, existiendo también causas adquiridas: tóxicas, inflamatorias, inmunomediadas, metabólicas, carenciales. Pueden presentarse en forma aislada o formando parte de un compromiso multisistémico.
Las ENM individualmente son enfermedades raras por su prevalencia e incidencia, pero en su conjunto afectan a un porcentaje significativo de la población.
Debido a la complejidad y el número creciente de estas enfermedades, anualmente se publica un resumen actualizado de las enfermedades musculares monogénicas con alteración primaria del genoma nuclear. A la publicación de 2017 donde se identifican 884 enfermedades y 492 genes, en 2018 se suman 8 variantes fenotípicas y 28 genes1.
Las ENM pueden manifestarse a cualquier edad, desde el nacimiento a la edad adulta. La manifestación clínica más común es la pérdida de fuerzas que puede ser progresiva o intermitente. Pueden presentar fatigabilidad, atrofia muscular, miotonía, calambres, compromiso cardíaco, alteraciones sensitivas o autonómicas. La debilidad determina la aparición de problemas secundarios, incluyendo ortopédicos (deformidades articulares), respiratorios, deglutorios que generan discapacidad y en casos severos pueden llevar a la muerte.
Algunas de las ENM más frecuentes son (Esquema 1):
- -
Distrofia muscular de Duchenne-Becker (DMD-DMB) es una enfermedad hereditaria autosómica recesiva ligada al cromosoma X, causada por alteración del gen de la distrofina. Existe debilidad en cintura de miembros, progresiva que lleva a la pérdida de la marcha. Las complicaciones incluyen alteraciones cardiorespiratorias y ortopédicas. La historia natural de la enfermedad ha cambiado por la terapia corticoidea2.
- -
Atrofia muscular espinal (AME) ligada al cromosoma 5 se presenta con un espectro clínico amplio que va desde la tipo 0 neonatal más severa a la tipo 4 del adulto. Las nuevas terapéuticas (ej: oligonucleótidos antisentido) pueden modificar la evolución de la enfermedad.
- -
Distrofia miotónica de Steinert es una enfermedad hereditaria con compromiso multisistémico, frecuente en la edad adulta con afectación cardíaca y respiratoria.

Diagrama de las enfermedades neuromusculares más frecuentes según la topografía de la afectación la unidad motora
Modificado de Hammer GD, McPhee SJ: Fisiopatología de la enfermedad 7e: www.accessmedicina.com
Si bien es cierto, que a la fecha no existe un tratamiento curativo para muchas de las ENM, en especial para las hereditarias, es incorrecto decir que estas enfermedades no tienen tratamiento, especialmente en este momento con la aparición de nuevas terapéuticas para algunas de ellas.
Las estrategias terapéuticas son cada vez más complejas y específicas por lo que requieren de un abordaje en los distintos niveles de atención en salud y rehabilitación (RHB).
Estos pacientes deben recibir tratamiento de RHB, de prevención de eventos cardiovasculares graves o letales, de medidas de soporte ventilatorio y nutritivo teniendo en cuenta la visión del paciente y su familia.
En este artículo abordaremos las enfermedades que afectan sobre todo la edad pediátrica.
ENFERMEDADES NEUROMUSCULARES Y SUS COMPLICACIONESManifestaciones del aparato locomotorLa debilidad es el síntoma principal y más característico de las ENM. Generalmente simétrica, en la mayoría de los casos es progresiva y condiciona gran parte de las complicaciones.
El grado de afectación varía de acuerdo a la patología, las de presentación precoz como AME tipo 1 o 2 graves, algunas distrofias musculares congénitas (DMC) y miopatías congénitas, presentan una acentuada debilidad desde el nacimiento.
Las miopatías congénitas presentan compromiso de músculos faciales, la debilidad es global a predominio proximal con evolución lentamente progresiva. En las DMC predomina en músculos faciales, masticatorios, cervicales, músculos paravertebrales y sectores proximales de los miembros.
Las retracciones tendino-musculares y deformidades articulares son frecuentes, debidas a disbalances musculares, posturas mantenidas, largos períodos de inmovilización y cambios fibróticos en el músculo. Generan mayor limitación funcional, por ejemplo la deformidad en equino varo puede acelerar la pérdida de la marcha en portadores de DMD.
Según la patología y la etapa evolutiva de la misma podemos analizar algunas presentaciones características:
- •
En el niño con DMD en etapa ambulatoria, existen retracciones del tendón de Aquiles y tensor de la fascia lata. Al progresar la enfermedad con pérdida de la marcha, aumentan las retracciones en miembros inferiores (flexores de cadera, rodillas) y se evidencian retracciones de miembros superiores.
- •
Las DMC se presentan retráctiles desde la etapa neonatal, en la evolución se agravan en forma progresiva, destacando por el compromiso funcional que generan: las retracciones de extensores del cuello y de la musculatura facial.
- •
En patología por colágeno VI, siendo más frecuente en distrofia muscular de Ullrich, se evidencia la aparición de importantes retracciones de codos y se suma a la hiperextensibilidad distal de dedos, la retracción de flexores de dedos. Tortícolis y retracciones distales de miembros inferiores son frecuentes al nacimiento.
- •
En el fenotipo de Emery Dreifuss (emerina, lámina A/C) existen retracciones de codos, aquileas y de extensores de cuello.
- •
La presentación de espina rígida por compromiso de los músculos paravertebrales se observa por ejemplo en las selenoproteinopatías.
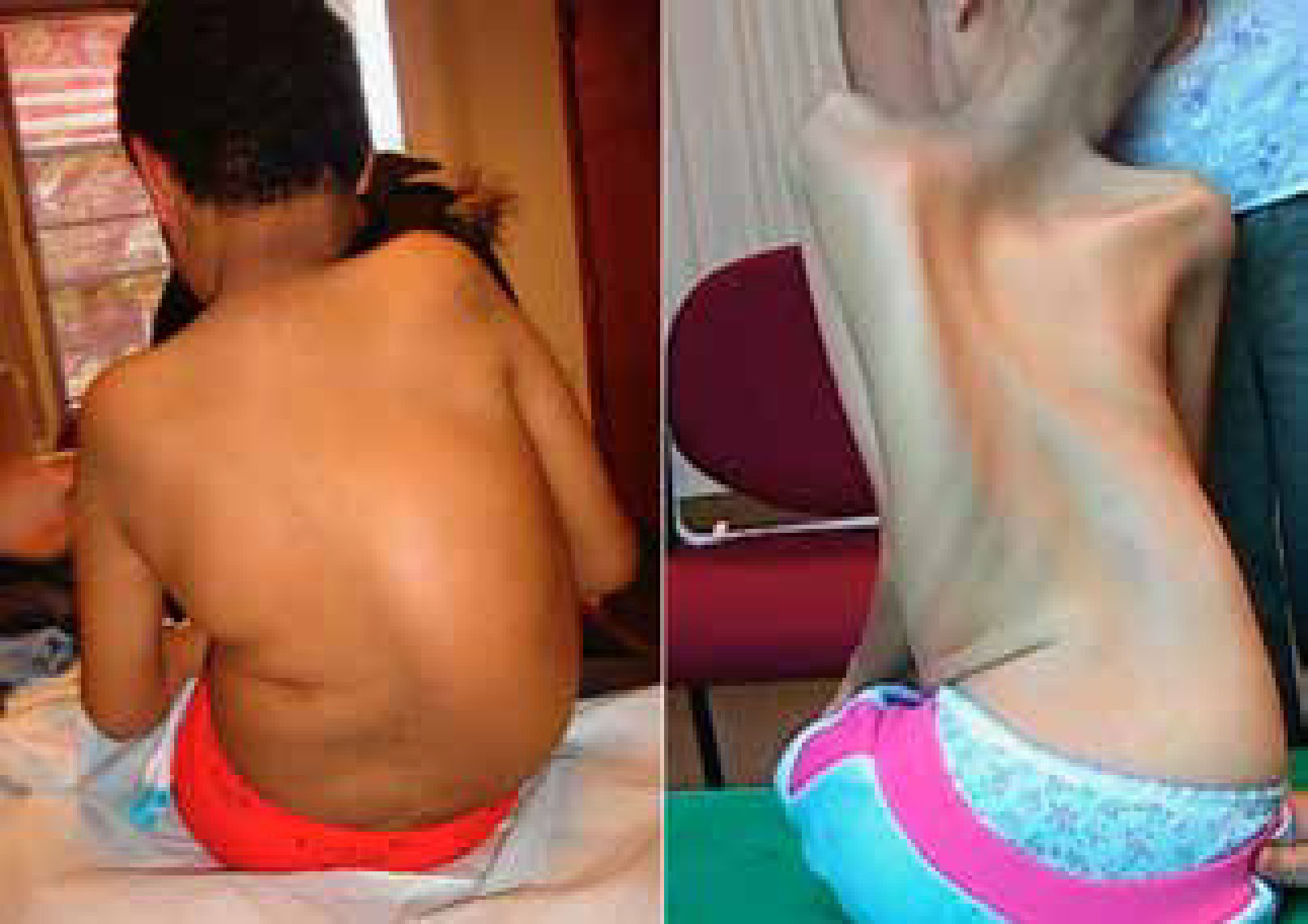
La escoliosis es generalmente flexible, producida por hipotonía y debilidad muscular. En algunos casos es una complicación rápidamente progresiva que genera inestabilidad postural, dolor y agravamiento del compromiso respiratorio.
En los pacientes con AME tipo 2 la cifoescoliosis es muy común y puede presentarse ya en el primer año de vida en las formas más graves3. A su vez en esta patología, la presencia de escoliosis severa, retracciones y aumento brusco de peso se ha visto relacionado con un empeoramiento rápido de la enfermedad4.
En las patologías de presentación neonatal como DMC y Enfermedad de Steinert congénito, la frecuencia y edad de aparición de la escoliosis es variable.
En DMD la escoliosis suele desarrollarse luego de la pérdida de la marcha. Se cita que hasta el 90% de los pacientes sin tratamiento corticoideo llegan a desarrollarla5.
Otras patologías neuromusculares pueden desarrollar escoliosis como por ejemplo las distrofias musculares de cintura de miembros (LGMD) y las neuropatías sensitivo motoras hereditarias (HMSN) aun sin perder la capacidad de marcha. (Figura 1).
")
La luxación de cadera es muy frecuente en AME tipo 2 y DMC con ausencia de marcha. Existe un déficit en la formación del acetábulo, que con la falta de carga y la debilidad muscular favorece la subluxación. Raramente son causa de dolor.
Las fracturas son otra complicación de estas patologías cuyo mecanismo tiene como base la falta o escasa motilidad. En la mayoría de los casos se relacionan con caídas pero pueden observarse por traumatismos mínimos. En DMD esta agravado por el tratamiento con corticoides. Publicaciones citan que hasta el 21% de pacientes habían presentado una fractura. En este grupo, el 50% de los pacientes mantenían la marcha6. En un trabajo que incluyó 65 pacientes con AME mostró que el 20% había presentado por lo menos una fractura7.
Otro mecanismo que se ha visto implicado en la etiología de las fracturas en AME es que el gen SMN1 participaría en el metabolismo fosfo-cálcico8.
El dolor es un síntoma frecuente que debe ser pesquisado en el interrogatorio y valorado por la escala análogo-visual del dolor.
Un trabajo publicado en 2015 sobre dolor en adolescentes portadores de AME y DMD encontró que el 69% había presentado dolor en los últimos 3 meses y que el 50% de la población refería dolor crónico9.
No abordaremos en este artículo el dolor neuropático, característico de las HMSN y que merece especial atención y tratamiento específico.
COMPROMISO RESPIRATORIOEn las ENM la debilidad del diafragma, los músculos intercostales y accesorios producen un defecto restrictivo y una insuficiencia respiratoria que puede llevar a la hipercapnia.
La valoración respiratoria forma parte del seguimiento de estos pacientes desde el diagnóstico o cuando se sospecha patología periférica, para detectar hipoventilación por compromiso respiratorio precoz.
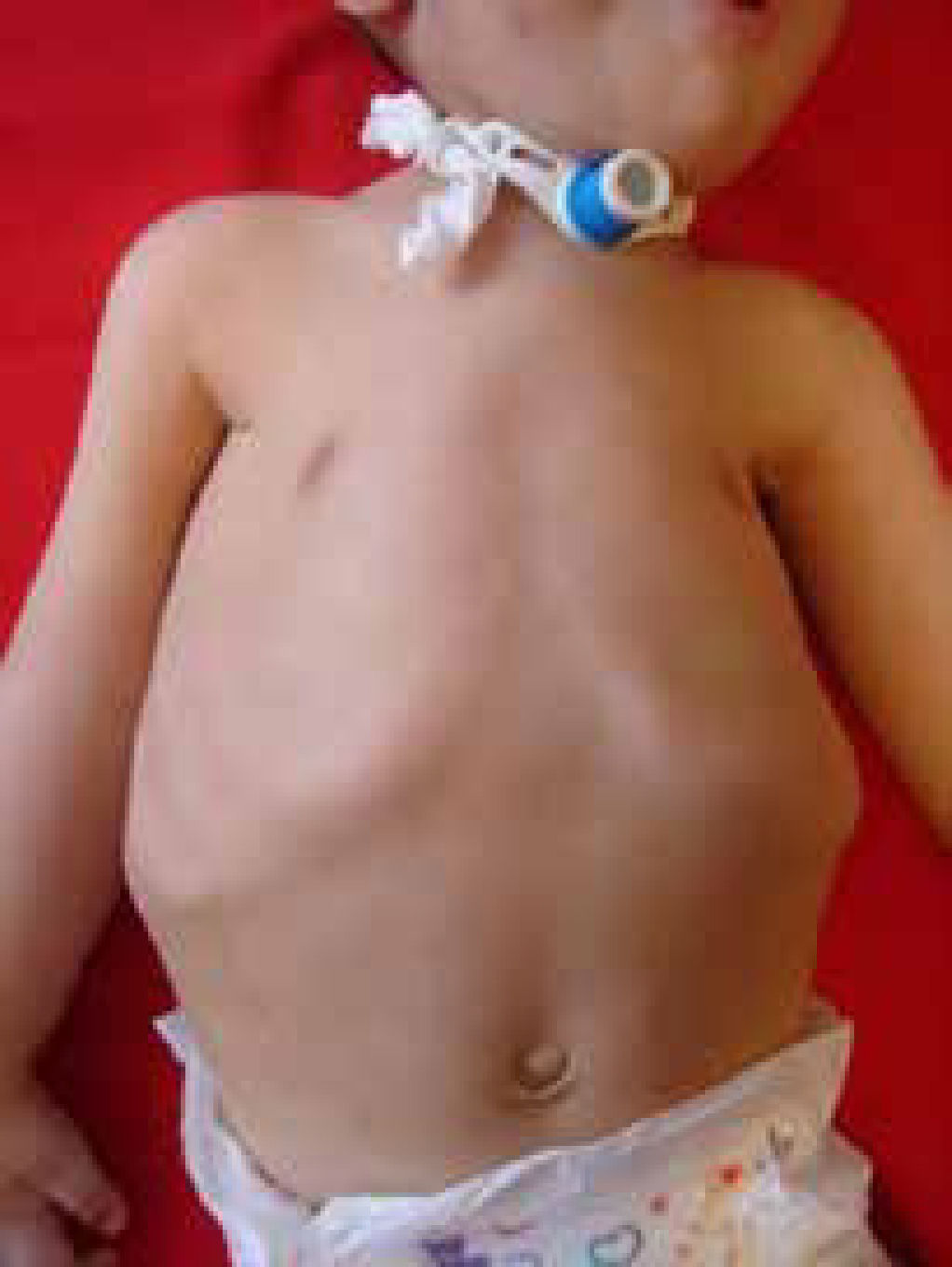
En los lactantes se monitoriza la frecuencia respiratoria, el patrón respiratorio, respiración paradójica, la forma del tórax y la eficacia de la tos. Se debe realizar una polisomnografía y/o una oximetría de pulso con gasometría matinal aunque es de baja sensibilidad10. (Figura 2).

En niños colaboradores, la Asociación Americana de Tórax aconseja en DMD una primera evaluación con espirometría forzada a partir de los 5 a 6 años11.
La función pulmonar muestra habitualmente un patrón restrictivo con disminución de la capacidad vital (CV), capacidad vital forzada (CVF), la capacidad pulmonar total (CPT) y la capacidad residual funcional (CRF), preservando relativamente el volumen espiratorio forzado del primer segundo (VEF1) y el índice VEF1/CVF.
Se debe realizar la espirometría en posición erguida y en decúbito dorsal para valorar el compromiso diafragmático12.
La disminución del esfuerzo respiratorio y del tono de la musculatura de la vía aérea que se produce durante el sueño fisiológico (sueño REM) se ve acentuada en estos pacientes produciendo hipoventilación y síndrome de apneas-hipopneas obstructivas del sueño. Los estudios de sueño tienen diferentes niveles de complejidad, la oximetría de pulso durante la noche es una herramienta útil para detectar hipoventilación nocturna y apneas obstructivas en el sueño. Todos los pacientes con oximetría anormal deben someterse a monitorización transcutáneo de pCo2 o polisomnografía13.
El mejor predictor de hipoventilación nocturna es la CVF, por lo que se recomienda realizar estudios de sueño con valores de CVF inferiores a 60%11. Las selenoproteinopatías y enfermedad de Steinert pueden presentar hipoventilación en el sueño con CV conservada.
Las pruebas de presión inspiratoria máxima (PIM) y presión espiratoria máxima (PEM) no proporcionan mayor información que la CVF13.
La evaluación de la tos se realiza determinando el pico flujo de la tos (PFT). En adultos cifras inferiores a 270 l/min indican deterioro en la capacidad para eliminar secreciones y establecen la necesidad de aplicar técnicas de tos asistida. En niños se ha comprobado que un PFT <160 l/min predice exacerbaciones pulmonares graves14.
El inicio electivo del soporte ventilatorio cambia la evolución y el pronóstico de los pacientes, ya que ha mostrado mejorar los gases arteriales, estabilizar las manifestaciones clínicas respiratorias, reducir las necesidades de hospitalización, incrementa el bienestar y la supervivencia, impactando en la calidad de vida.
La Ventilación Mecánica no Invasiva (VNI) es uno de los procedimientos fundamentales en el manejo de los pacientes con Esclerosis Lateral Amiotrofica (ELA), mejorando la calidad de vida y la supervivencia requiriendo en la evolución considerar formas de ventilación mecánica a través de traqueotomía15. Se sugiere vacuna influenza anual y neumococo11,16.
COMPROMISO CARDÍACOMuchas de las proteínas involucradas en enfermedades del músculo esquelético afectan el músculo cardíaco. Se diferencian varios grupos de proteínas: de la unidad contráctil (complejo actina-miosina, las proteínas de enlace y las proteínas con interacción con el ATP), proteínas de anclaje del citoesqueleto a la membrana nuclear (lámina A/C y emerina) y a la membrana celular (complejo distrofina, sarcoglicanos, calpaína), canales iónicos de membrana y proteínas con función enzimática o metabólica.
Las alteraciones de la unidad contráctil se manifiestan como cardiomiopatías hipertróficas, mientras que las del citoesqueleto como cardiomiopatías dilatadas17. Las formas de presentación son: miocardiopatías hipertróficas, dilatadas y arritmias.
Nos referiremos a algunas de las ENM con compromiso cardíaco:
Distrofinopatías: Las complicaciones cardiovasculares son la principal causa de morbimortalidad de la DMD11. Se presentan como cardiomiopatía dilatada-hipertrófica. Los síntomas y signos de falla cardíaca en pacientes no ambuladores son sutiles y por lo tanto, no diagnosticados, deben ser sospechados y evaluados precozmente. En la DMB el compromiso cardíaco es similar al observado en la DMD, la cardiopatía es más evidente por el menor compromiso motor. Existe un 15% de prevalencia de cardiopatía antes de los 16 años y 73% a los 40 años18.
Las portadoras de la alteración genética de la DMD pueden padecer alteraciones cardíacas hasta en un 47%, en hallazgos imagenológicos19.
Se plantea realizar evaluación cardiológica con ecocardiograma (<6-7 años) o RNM cardíaca (>6-7 años) al diagnóstico de la enfermedad, luego anualmente o frente a cirugía. El inicio de tratamiento con inhibidores de la enzima convertidora de angiotensina (IECA) en niños con DMD a los 10 años de edad con función cardiaca normal podría mejorar los posibles trastornos cardíacos a largo plazo11.
Los adultos con distrofia miotónica de Steinert pueden presentar alteraciones en el ecocardiograma, pero, no siempre se expresan clínicamente. Se ha observado cardiomiopatía hipertrófica y bloqueos de la conducción aurículo-ventricular (AV) que requieren colocación de marcapasos.
La afectación cardíaca es frecuente en varios subtipos de las LGMD: LGMD 2A (calpaína), LGMD 2J (titina), LGMD 2I (futukin related protein), LGMD 1C (lamina A/C).
En la enfermedad de Emery Dreifuss (emerina, lamina A/C) son frecuentes la cardiomiopatía dilatada y los bloqueos AV. Se han descripto casos asociados a alteraciones de lamina A/C que se presentan como muerte súbita.
En la Distrofia Facio Escápulo Humeral se observan alteraciones como disfunción del nodo sinusal y bloqueos AV.
Las miopatías metabólicas (errores del metabolismo de la carnitina, beta oxidación de ácidos grasos) presentan cardiomiopatía dilatada casi constante y arritmias ventriculares graves. Las glucogenosis y algunas enfermedades mitocondriales se presentan con cardiomiopatía hipertrófica.
En AME la presencia de cardiopatías es poco común, se han identificado defectos cardíacos congénitos en pacientes con forma neonatal severa tipo 016.
TRASTORNOS DEL APARATO DIGESTIVO, COMPROMISO DEL ESTADO NUTRICIONAL Y ALTERACIONES METABÓLICASLa debilidad muscular dificulta la masticación y deglución generando disfagia. Puede asociarse a disfunción bulbar (AME, ELA) o macroglosia (DMD).
Los síntomas de la disfagia son tos, atragantamiento, fatiga y sudoración.
La dificultad para comer se ve agravada por la limitación de la apertura bucal y debilidad de las extremidades superiores, requiriendo tratamiento fonoaudiológico en las áreas de deglución y la comunicación.
El reflujo gastroesofágico repercute sobre aspectos nutricionales y respiratorios secundarios a episodios de aspiración. La enfermedad respiratoria crónica con infecciones intercurrentes lleva a un mayor consumo energético y compromiso del estado nutricional.
Existen otros trastornos gastrointestinales como alteración de la motilidad con distensión abdominal comprometiendo la dinámica respiratoria, retraso del vaciado gástrico y estreñimiento crónico.
Estas complicaciones suelen estar presentes en la mayoría de las ENM, son subdiagnosticadas y deben ser buscadas en forma rutinaria requiriendo abordaje precoz. Las alteraciones nutricionales determinan compromiso del desarrollo pondo estatural que son precoces y graves en AME 1 y DMC. En cambio, los portadores de DMD ambuladores suelen tener sobrepeso-obesidad y baja talla si están en corticoterapia. En etapas avanzadas de DMD hay desnutrición de causa multifactorial.
Consideraciones a tener en cuenta en algunas enfermedades:
- -
AME tipo 1 se sugiere realizar una evaluación precoz con videofluoroscopía al momento del diagnóstico, en AME tipo 2 se realiza frente a la sospecha clínica de los síntomas de disfagia-aspiración. En AME tipo 3 los trastornos deglutorios son raros.
De existir alteraciones de la deglución o falla del crecimiento en AME tipo 1 o 2 se plantea en forma transitoria sonda nasogástrica hasta decisión de gastrostomía (con o sin funduplicatura de Nissen). Se debe controlar el metabolismo dado que pueden presentar baja tolerancia a las grasas, hiperlipidemia y alteración del metabolismo de la glucosa, acidosis metabólica temprana y deshidratación20.
- -
DMD el trastorno de la deglución puede ser precoz aún sin síntomas evidentes. Se controla clínicamente cada 6 meses. Frente a disfagia moderada-severa, aspiración, pérdida de peso o desnutrición se sugiere la gastrostomía. Compromiso del crecimiento, retardo puberal e insuficiencia suprarrenal son las complicaciones endocrinológicas de DMD y del tratamiento corticoideo que deben ser controladas21.
- -
Distrofia miotónica (Steinert) existe trastorno de la deglución (fase oral) por debilidad de maseteros, atrofia de musculatura facial y deformaciones de mandíbula.
- -
DMC el trastorno de la deglución por debilidad de músculos faciales, mala oclusión dentaria asociado a reflujo gastroesofágico compromete el estado nutricional.
- -
ELA el estado nutricional y el peso son importantes factores de predicción de supervivencia. Frente a la desnutrición se plantea la realización de una gastrostomía, podría mejorar la supervivencia (sin evidencias definitivas)15.
Generalmente los pacientes con ENM tienen un nivel cognitivo conservado.
En DMD han sido reportados: déficit intelectual (17-27%), dificultades de aprendizaje (26%), trastorno del espectro autista (15%), trastorno por déficit atencional e hiperactividad (32%) y ansiedad (27%)22. En Distrofia miotónica de Steinert existe un alto porcentaje de déficit intelectual. Un estudio en niños y adolescentes con AME mostró un nivel de inteligencia comparable con el grupo control, encontrando en los adolescentes un nivel cognitivo más elevado, lo que sugiere que los mismos desarrollan mecanismos compensadores del compromiso motor23.
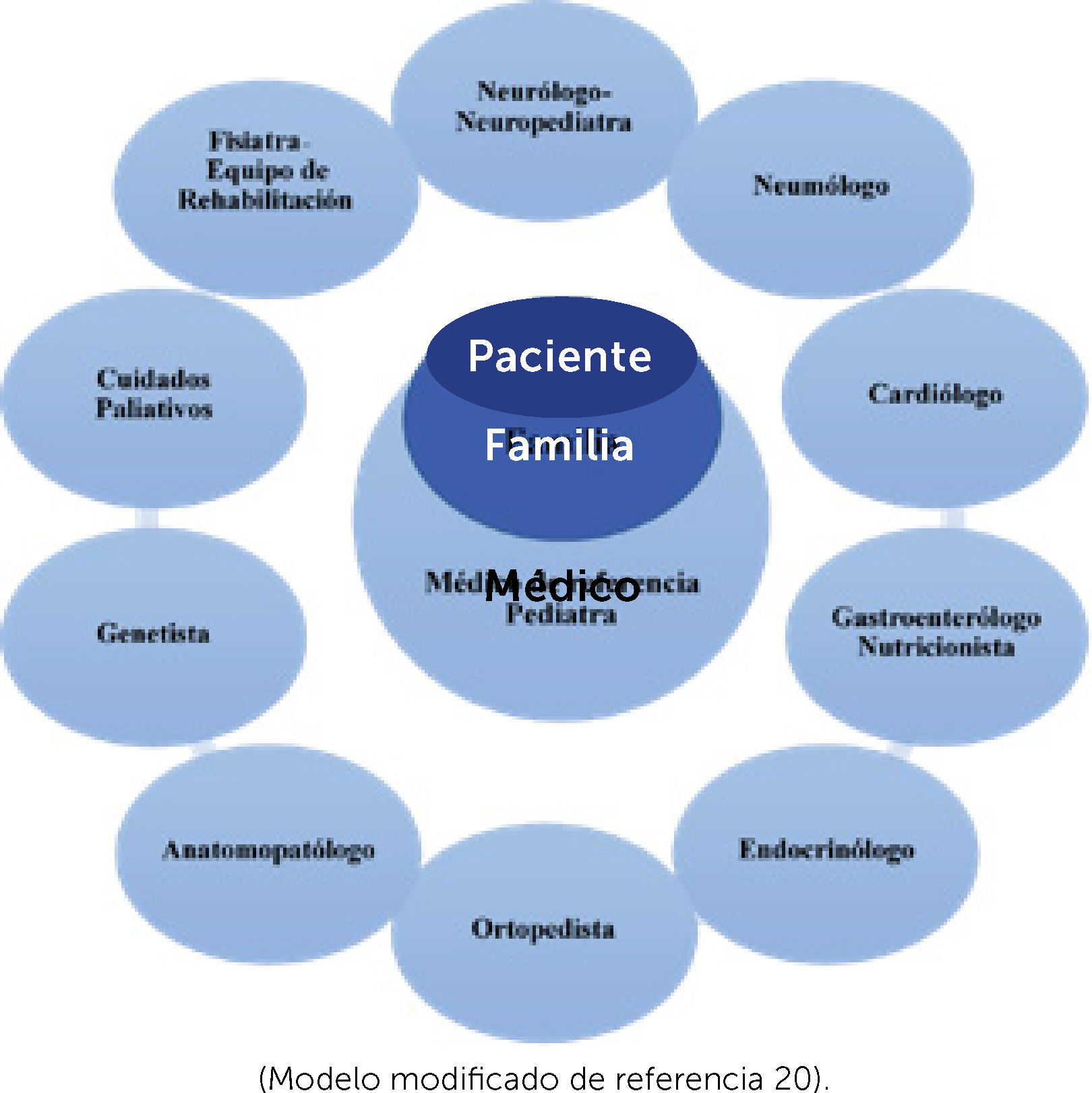
TRATAMIENTO DE HABILITACIÓN-REHABILITACIÓNLa gran variedad de ENM con evolución y pronósticos diferentes, la dificultad en el diagnóstico y los avances terapéuticos exigen la intervención de múltiples disciplinas médicas y de un equipo interdisciplinario en rehabilitación coordinado por médico fisiatra, conformado por licenciados en trabajo social, psicología, kinesiología, terapia ocupacional, fonoaudiología, enfermería y pedagogía. De acuerdo a la necesidad de cada caso se sumaran otros profesionales (odontólogo, licenciados en órtesis, ingenieros biomédicos, entre otros).
El equipo de cuidado paliativo toma un rol importante en todas las etapas de la vida de estos enfermos: en el momento del diagnóstico, frente a decisiones del tratamiento (gastrostomía VNI o AVM), eventos agudos y acompañamiento en etapa final de la vida. (Esquema 2).
.")
Diagrama equipo multidisciplinario de diagnóstico y tratamiento de enfermos neuromusculares
(Modelo modificado de referencia 20).
La valoración del usuario, tomando como marco conceptual a la Clasificación Internacional del Funcionamiento, de la Discapacidad y de la Salud (CIF), debe abarcar todos los factores que determinan la discapacidad realizando el control, prevención y tratamiento de los complicaciones que van apareciendo en la evolución, con el objetivo general de lograr la mayor participación que el estadio de la enfermedad permita, promoviendo la inclusión de la persona en todos los ámbitos. (Esquema 3).

Una vez realizada esta evaluación, se pautará el programa de rehabilitación y se definirá el seguimiento. Destacaremos los objetivos específicos y mencionaremos algunos de los lineamientos terapéuticos fundamentales:
- 1.
Mantener o mejorar cuando es posible la fuerza muscular: Rol del ejercicio en las ENM. Sigue siendo controvertido en algunas patologías como las distrofias musculares dado que no es claro su efecto en la degeneración muscular. En DMD-B se reitera la necesidad de evitar la actividad muscular excéntrica y el ejercicio de alta resistencia. Se sugiere actividad aeróbica submáxima evitando sobreesfuerzo, permitiendo adecuado descanso, ejercicios en piscina y bicicleta (en pacientes mayores utilizarla en forma asistida o con movimientos asistidos por robótica)21.
En AME debe estimularse el ejercicio y fomentar la actividad muscular. Para no ambulantes se recomiendan actividades en piscina, ejercicios concéntricos-excéntricos, ejercicios aeróbicos y de acondicionamiento general. En ambulantes se agregan ejercicios de equilibrio20.
En las polineuropatías subagudas y crónicas hereditarias, el programa de fortalecimiento muscular se plantea de acuerdo a la tasación muscular.
- 2.
Prevenir complicaciones secundarias determinadas por la debilidad muscular: las retracciones tendinomusculares, la escoliosis y favorecer el desarrollo tóraco-pulmonar (en los casos en que la enfermedad se manifiesta desde las primeras etapas de la vida), abordaje del dolor.
Prevención de complicaciones ortopédicas:
Retracciones tendinomusculares y deformidades articulares, priorizando en niños que crecen con una ENM. El programa incluye:
- A.
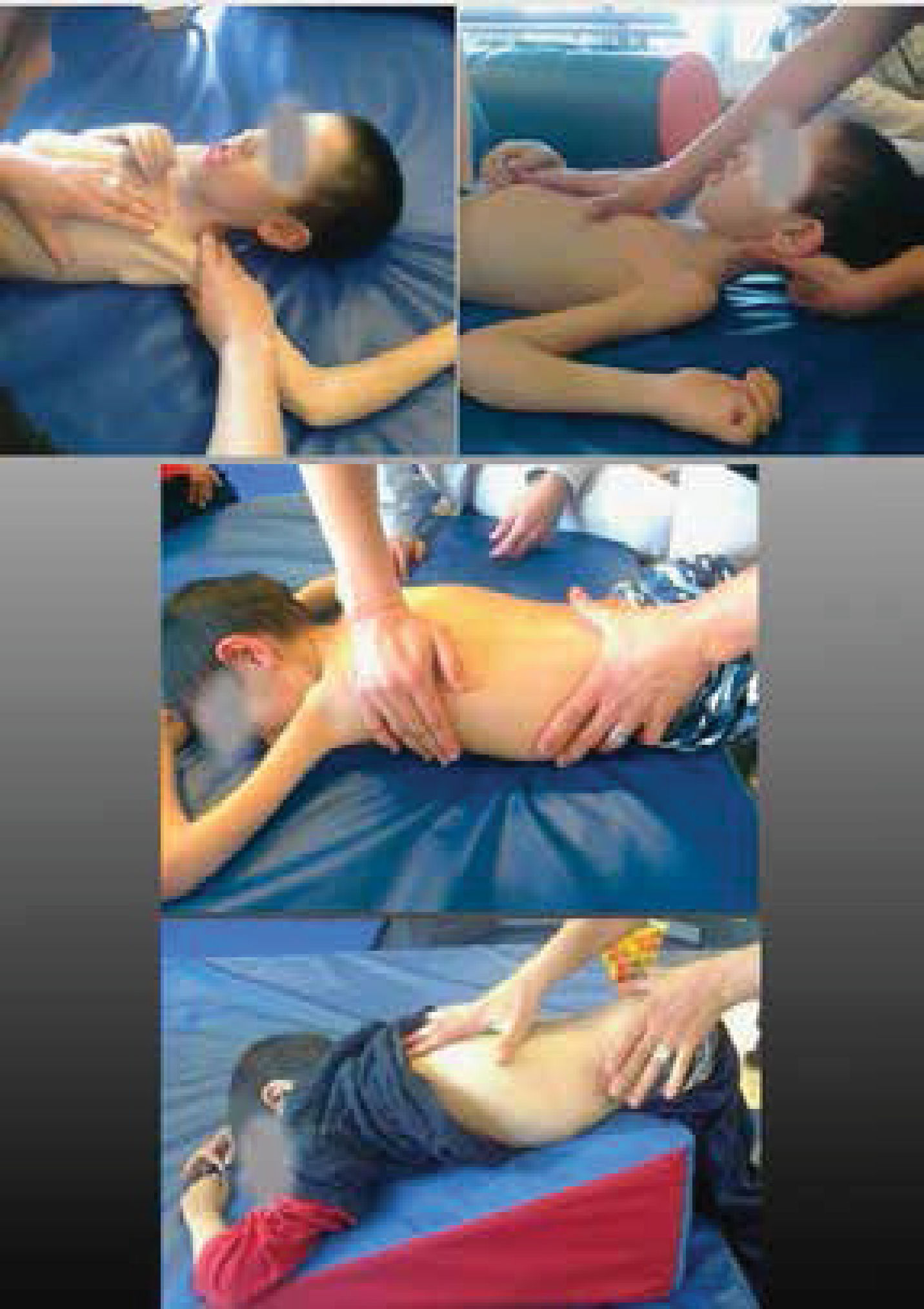
Elongaciones pasivas realizadas en forma precoz e intensiva; orientando al cuidador en qué articulaciones priorizar de acuerdo a la evaluación y la patología. En la última revisión sobre DMD se sugiere programas de elongaciones en domicilio de 4 a 6 veces por semana21. Expertos en AME sugieren para los niños no sedentes una frecuencia mínima de 3 a 5 veces por semana y para los sedentes 5 a 7 veces por semana20. (Figura 3).
- B.
Tecnología asistiva para posicionamiento y correcta alineación corporal:
- •
Uso de ortesis, ej: en el niño con DMD en etapa de ambulación se indica férulas antiequino de uso nocturno y, una vez que pierde la marcha continua, con uso diurno para prevenir la deformidad en equinovaro que de progresar hace imposible el uso de calzados.
- •
Almohadones preformados y lechos para el posicionamiento durante el sueño, evitando en niños la actitud en batracio que puede generar luxación anterior de cadera y acortamiento del tensor de la fascia lata.
- •
Bipedestadores estáticos o motorizados, mientras las retracciones no lo impidan. La bipedestación ayuda a prevenir complicaciones ortopédicas, prevenir osteoporosis, influye positivamente en la función respiratoria y digestiva.
- •
Sillas fijas para actividades en la escuela, liceo, en talleres o de uso en el hogar/accesorios para silla de ruedas que permitan mejor posturación.
- •
Abordaje de la escoliosis- No hay consenso en relación al beneficio de las órtesis de tronco en niños con ENM, no logran evitar la progresión de las curva. En AME sedentes se recomienda su uso en escoliosis con curva mayor de 15°-20° especialmente en niños con potencial de crecimiento20. En DMD no recomiendan su uso. En curvas mayores de 20 grados se plantea valoración por ortopedista11.
Se continúa indicando órtesis de tronco con un objetivo funcional, en estos casos se debe vigilar que no limite más la función respiratoria (realizar ventana anterior y controlar con funcional respiratorio).
En AME la decisión de la instrumentación quirúrgica de la columna depende de la magnitud de la curva (ángulo de Cobbs >50°) y del rango de progresión (más de 10° por año). Se tendrán en cuenta además otros factores como la función respiratoria, deformidad de la caja torácica y el impacto funcional. Hay consenso en no realizar cirugías antes de los 4 años. En los últimos años se han desarrollado nuevas técnicas quirúrgicas para aplicar en niños con esqueleto inmaduro, antes de la fusión espinal definitiva, ej: Growing-rods20.
Con respecto al abordaje de la luxación y subluxación de cadera, el último consenso AME 2018 sugiere cirugía en caso de dolor.
Prevención de complicaciones respiratorias
Los objetivos del tratamiento estarán destinados a:
- A.
Mantener la elasticidad de los pulmones, la caja torácica y promover su normal crecimiento realizando: movilizaciones manuales (especialmente en musculatura intercostal y tronco), hiperinsuflaciones con ambú o equipos mecánicos y respiración glosofaríngea.
- B.
Facilitar el “clearence” de la vía aérea: posturas de drenaje, asistencia manual y asistencia mecánica de la tos (insuflaciones-exuflaciones, terapia con chaleco).
Existen estudios que confirman que esta intervención de insuflaciones/exuflaciones realizadas en forma regular durante años enlentecen la pérdida de la función y mejoran la capacidad vital, reduciendo las hospitalizaciones y mejorando la calidad de vida24.
- A.
- 3.
Mantener el mayor tiempo posible la autonomía al realizar las actividades de la vida diaria (AVD) y promover la participación, teniendo en cuenta factores personales y ambientales, impactando de esta forma en la calidad de vida de los individuos. Todas las disciplinas que conforman el equipo estarán enfocadas en este objetivo. Se realizara un abordaje de la dependencia del usuario incluyendo el asesoramiento a cuidadores. Se evaluará la prescripción de recursos existentes de tecnología asistiva (alta o baja tecnología) más adecuados para la comunicación y/o movilidad. Se tendrá en cuenta para ello: edad, evolución de la patología, contexto (escuela, comunidad, trabajo) y realidad de cada familia.

Se cuenta con diferentes dispositivos de tecnología asistida para AVD, productos adquiridos comercialmente, modificados o hechos a medida como pueden ser un cubierto engrosado, dispositivo de acceso a computadoras, sillas de baño o grúas para facilitar las transferencias.
Para promover la movilidad independiente existen diferentes tipos de silla de ruedas: de autopropulsión y motorizadas (SRM) con accesorios de alta complejidad que permiten mayor independencia a personas con grandes limitaciones motoras.
Teniendo en cuenta el nivel de funcionamiento cognitivo, se recomienda el uso de dispositivos motorizados para movilidad desde temprana edad (en niños con AME tipo 2 se prescribe SRM a partir de los dos años).
La adaptación del entorno facilita la movilidad, accesibilidad, autonomía y tarea del cuidador. Se debe realizar valoración de los ambientes que frecuenta el usuario (vivienda, institución educativa, trabajo, entre otros) con el objetivo de intervenir buscando un entorno lo más accesible posible (ej, sistemas de control: domótica).
La tecnología asistiva pierde su razón de ser si no se acompaña de un medio accesible para todos sin necesidad de adaptaciones: diseño universal.
La educación es un derecho debiéndose garantizar el acceso a la misma a todas las personas. Para los niños con ENM se debe elaborar un plan de intervención personalizado que contemple: modo de escolarización, necesidad de asistente personal, tipo de dispositivos de tecnología asistiva que requiera y necesidad de ajustes razonables. Crear un ambiente de aprendizaje inclusivo ayudará a todos los niños a aprender y desarrollar su potencial.
Destacamos la importancia del ocio y el tiempo libre: las actividades lúdico-deportivas son un medio adecuado para contribuir al desarrollo integral de las personas con discapacidad. Los espacios de ocio y de convivencia social deber ser accesibles.
Abordaje psicosocial:
El trabajo con patologías evolutivas, tiene un alto impacto emocional en todos los actores de este proceso. Estas enfermedades conllevan pérdidas progresivas, deterioro de la calidad de vida, aumento de limitaciones para desempeñarse en la vida diaria, necesidad de dependencia y enfrentan a todos en forma explícita e implícita al sentido de la vida y a la muerte.
El trabajo interdisciplinario es un factor de protección para el equipo de salud. Beneficia al usuario y su familia en múltiples sentidos. El rol del psicólogo es en primera instancia realizar un diagnóstico situacional, evaluar qué información han recibido, que han podido asimilar de ésta, que mecanismos de defensa ante la angustia van desarrollando y con qué recursos cuentan. En una segunda instancia se trabaja en el proceso de afrontamiento, para encontrar estrategias y recursos para vivir con la enfermedad.
El trabajador social elaborará junto a la persona y su familia (en la medida de lo posible) líneas de intervención que apunten a potenciar sus capacidades de desarrollo humano y su autonomía. Se trabaja en interacción con la comunidad, comprometiendo a los involucrados a realizar las adaptaciones necesarias para conseguir la participación efectiva en la vida social, potenciando el bienestar y goce de los derechos.
CONCLUSIONESLas ENM en su mayoría graves y progresivas, pueden generar una gran discapacidad con impacto a nivel personal, familiar, social, generando altos costos en salud.
En los últimos años ha mejorado la expectativa de vida de las personas con ENM. El manejo multidisciplinario y el programa de rehabilitación junto con los avances terapéuticos-tecnológicos han permitido mejorar la calidad de vida y participación.
AgradecimientosLos autores agradecen a las Lic. C. Castello, S. Meroni, B. Silveira De Andrade, I. Pommerenck del equipo de ENM del Centro de Rehabilitación Infantil Teletón-Uruguay; a los pacientes y sus familiares.
Declaración de interesesNo existen conflictos de interés respecto a este artículo. Las fotos han sido autorizadas para su publicación.