




La rabdomiólisis resulta de la destrucción del tejido muscular con liberación de sus componentes a la circulación sistémica, lo que puede ser potencialmente grave. Sus causas pueden ser adquiridas o hereditarias (genéticas-metabólicas), y según los mecanismos fisiopatológicos se han clasificado en causas metabólicas (falla en la producción de energía), estructurales (miopatías estructurales o distrofias), por alteración en la bomba de calcio y causas inflamatorias. Independiente de la causa de la rabdomiólisis, la vía final común es la injuria del sarcolema, ya sea por aumento del calcio intracelular o por falla de la producción de energía, lo que conduce a la necrosis de la fibra y, consecuentemente, la liberación a la circulación de electrolitos y proteínas intracelulares. Las anormalidades metabólicas más frecuentemente reportadas con rabdomiólisis son la deficiencia de Carnitina Palmitoil transferasa II (CPT II), defectos de glicólisis y recientemente se han descrito mutaciones en el gen de Lipina 1 (LPIN1). Ante un episodio de rabdomiólisis se deben descartar inicialmente trastornos adquiridos potencialmente tratables, para luego dar paso a estudios de miopatías estructurales o metabólicas. Respecto al manejo, en fase aguda tiene como objetivo preservar la función renal y restaurar anormalidades metabólicas, a través de un aporte precoz y adecuado de volumen, asegurando una adecuada diuresis, y en situaciones de acidosis o hiperkalemia extremas, debe considerarse la hemodiálisis o terapias de reemplazo renal continuas. Se debe tener en consideración, complicaciones graves en este cuadro clínico, como la coagulación intravascular diseminada y síndromes compartimentales que pueden requerir fasciotomías múltiples precoces. Específicamente, en mutaciones en gen de LPIN1, causantes de hasta el 50% de los episodios de rabdomiólisis en edad pediátrica, las estrategias terapéuticas deben ser dirigidas a la prevención y tratamiento precoz del catabolismo, a través del aporte de soluciones intravenosas con elevadas concentraciones de glucosa.
Rhabdomyolysis results from the destruction of muscle tissue and the release of its components into the systemic circulation, which can be potentially serious. Causes can be acquired or hereditary (genetic-metabolic), and according to the pathophysiological mechanisms have been classified into metabolic causes (failure in the production of energy), structural (structural myopathies or dystrophies), by abnormalities in the calcium pump and inflammatory causes. Regardless of the cause of rhabdomyolysis, the common final mechanism is the sarcolemma injury, either by increased intracellular calcium or by failure of energy production, which leads to fiber necrosis and, consequently, the release to the circulation of electrolytes and intracellular proteins. Metabolic abnormalities most frequently reported with rhabdomyolysis are Carnitine palmitoyltransferase II deficiency (CPT II), glycolysis defects, and mutations in the Lipin 1 gene (LPIN1), which have been described recently. Under an episode of rhabdomyolysis, potentially treatable acquired disorders should be ruled out initially, and then studies of structural or metabolic myopathies should be carried out. Regarding management, in acute phase aims to preserve renal function and restore metabolic abnormalities, through an early and adequate volume, ensuring adequate diuresis, and in situations of extreme acidosis or hyperkalemia, should be considered hemodialysis or therapies of continuous renal replacement. Consideration should be given to serious complications in this clinical picture, such as disseminated intravascular coagulation and compartment syndromes that may require early multiple fasciotomies. Specifically, in mutations in the gene of LPIN1, which cause up to 50% of episodes of rhabdomyolysis in the pediatric age, therapeutic strategies should be directed to the prevention and early treatment of catabolism, through the use of intravenous solutions with high concentrations of glucose.
La rabdomiólisis es el resultado de la rápida destrucción de las fibras musculares, con salida del contenido intracelular a la circulación sistémica, lo que es potencialmente tóxico1.
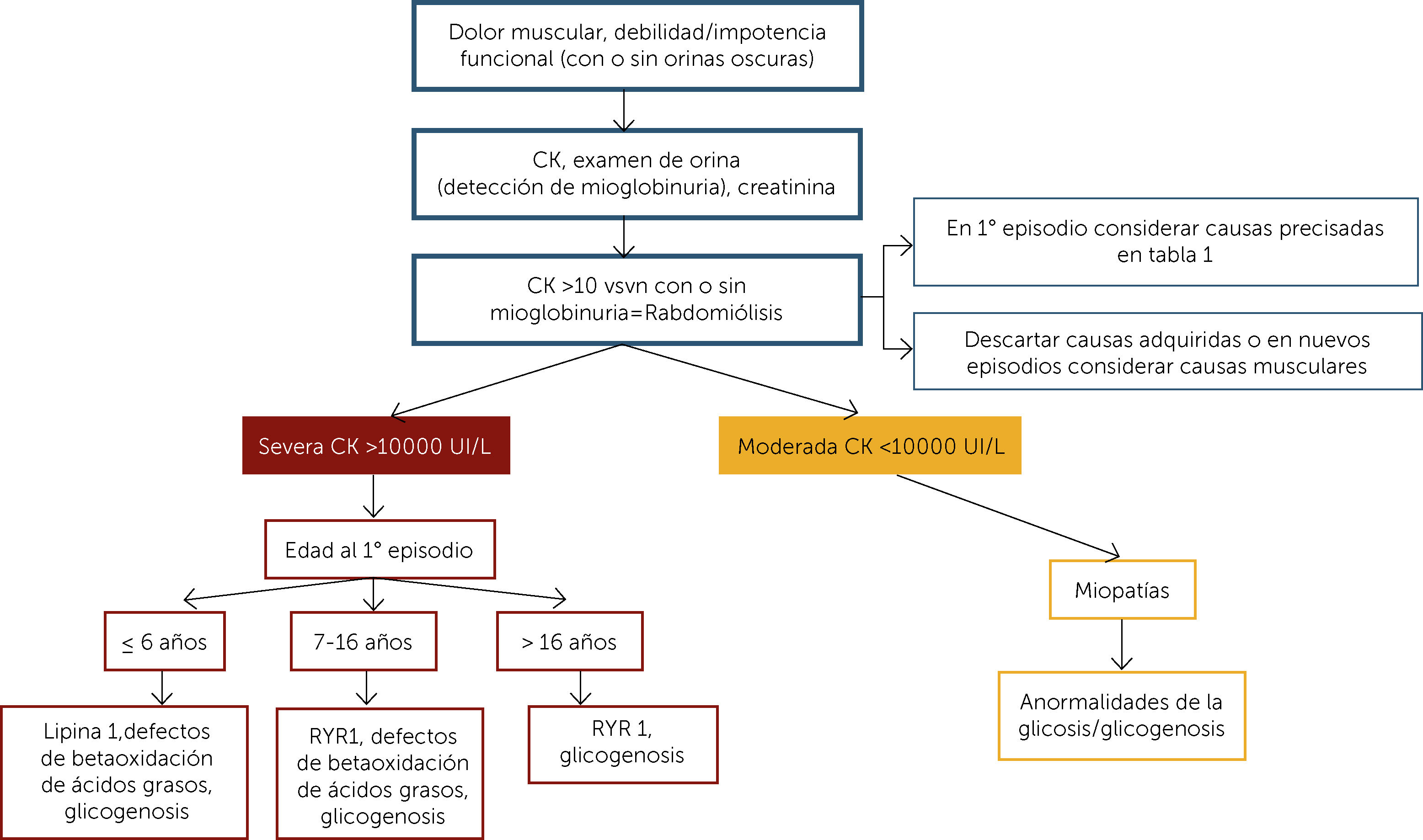
Se manifiesta por dolor muscular, debilidad e impotencia funcional, edema, orina de color café oscuro y elevación de la creatinkinasa (CK) sérica por sobre 10 veces el valor superior normal2.
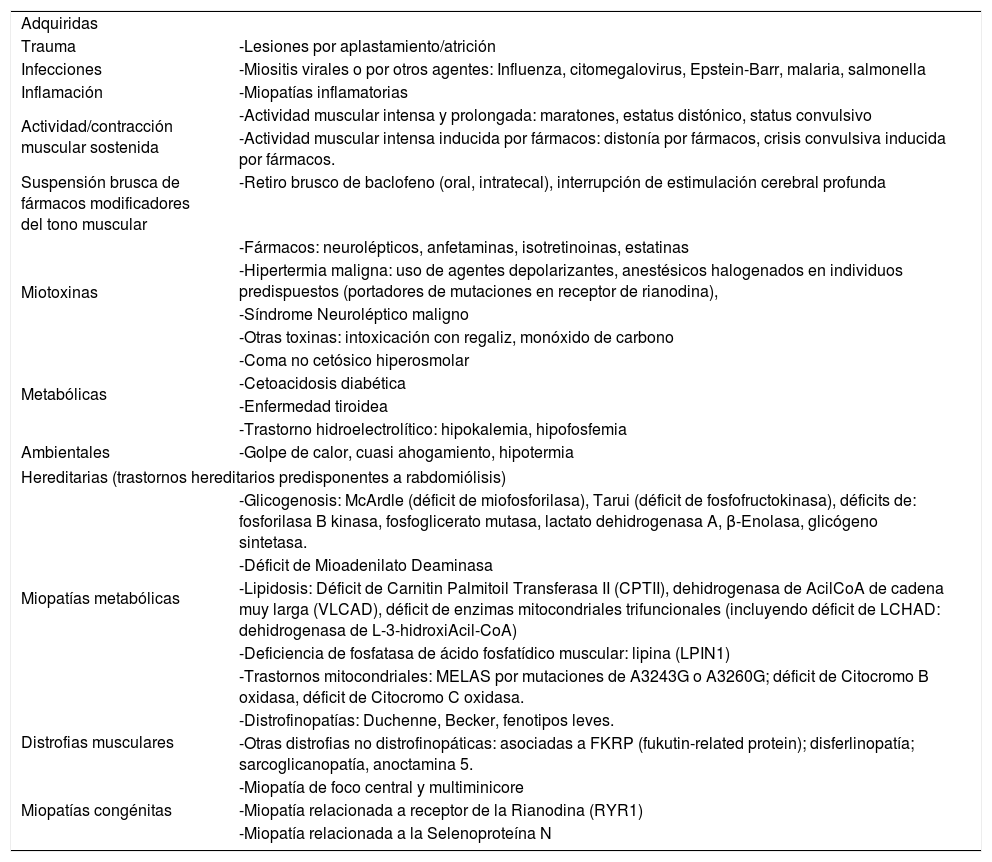
Sus causas pueden ser adquiridas (situaciones que determinan una injuria directa al sarcolema) o hereditarias (condiciones genéticamente determinadas que originan un trastorno metabólico o estructural que lleva a necrosis de fibras musculares)3, las que se presentan en la Tabla 1.
Causas de rabdomiólisis en niños
| Adquiridas | |
| Trauma | -Lesiones por aplastamiento/atrición |
| Infecciones | -Miositis virales o por otros agentes: Influenza, citomegalovirus, Epstein-Barr, malaria, salmonella |
| Inflamación | -Miopatías inflamatorias |
| Actividad/contracción muscular sostenida | -Actividad muscular intensa y prolongada: maratones, estatus distónico, status convulsivo |
| -Actividad muscular intensa inducida por fármacos: distonía por fármacos, crisis convulsiva inducida por fármacos. | |
| Suspensión brusca de fármacos modificadores del tono muscular | -Retiro brusco de baclofeno (oral, intratecal), interrupción de estimulación cerebral profunda |
| Miotoxinas | -Fármacos: neurolépticos, anfetaminas, isotretinoinas, estatinas |
| -Hipertermia maligna: uso de agentes depolarizantes, anestésicos halogenados en individuos predispuestos (portadores de mutaciones en receptor de rianodina), | |
| -Síndrome Neuroléptico maligno | |
| -Otras toxinas: intoxicación con regaliz, monóxido de carbono | |
| Metabólicas | -Coma no cetósico hiperosmolar |
| -Cetoacidosis diabética | |
| -Enfermedad tiroidea | |
| -Trastorno hidroelectrolítico: hipokalemia, hipofosfemia | |
| Ambientales | -Golpe de calor, cuasi ahogamiento, hipotermia |
| Hereditarias (trastornos hereditarios predisponentes a rabdomiólisis) | |
| Miopatías metabólicas | -Glicogenosis: McArdle (déficit de miofosforilasa), Tarui (déficit de fosfofructokinasa), déficits de: fosforilasa B kinasa, fosfoglicerato mutasa, lactato dehidrogenasa A, β-Enolasa, glicógeno sintetasa. |
| -Déficit de Mioadenilato Deaminasa | |
| -Lipidosis: Déficit de Carnitin Palmitoil Transferasa II (CPTII), dehidrogenasa de AcilCoA de cadena muy larga (VLCAD), déficit de enzimas mitocondriales trifuncionales (incluyendo déficit de LCHAD: dehidrogenasa de L-3-hidroxiAcil-CoA) | |
| -Deficiencia de fosfatasa de ácido fosfatídico muscular: lipina (LPIN1) | |
| -Trastornos mitocondriales: MELAS por mutaciones de A3243G o A3260G; déficit de Citocromo B oxidasa, déficit de Citocromo C oxidasa. | |
| Distrofias musculares | -Distrofinopatías: Duchenne, Becker, fenotipos leves. |
| -Otras distrofias no distrofinopáticas: asociadas a FKRP (fukutin-related protein); disferlinopatía; sarcoglicanopatía, anoctamina 5. | |
| Miopatías congénitas | -Miopatía de foco central y multiminicore |
| -Miopatía relacionada a receptor de la Rianodina (RYR1) | |
| -Miopatía relacionada a la Selenoproteína N | |
(Modificado de: Chan EK, Kornberg AJ, Ryan MM.A diagnostic approach to recurrent myalgia and rhabdomyolysis in children. Arch Dis Child. 2015;100(8):793–7).
Si bien las causas adquiridas dan cuenta de la mayoría de estos trastornos, algunas miopatías metabólicas presentan predisposición a rabdomiólisis, y pueden ser sospechadas desde esta situación clínica4. Se ha reportado que hasta un 10% de los casos de rabdomiólisis tienen una miopatía metabólica como causa subyacente, existiendo un alto riesgo de recurrencia5. Se consideran en este grupo, algunos trastornos como los defectos de metabolismo del glicógeno, de la beta-oxidación de los ácidos grados y de la fosforilación oxidativa mitocondrial4, ante los cuales es importante su sospecha clínica, su prevención y un manejo pronto y adecuado.
La severidad de la rabdomiólisis puede ir desde una elevación de CK en forma asintomática o con síntomas leves, hasta la falla renal aguda, arritmias cardíacas, síndrome compartimental, shock hipovolémico o coagulación intravascular diseminada2,5, de ahí la importancia de su sospecha diagnóstica y su adecuado manejo.
Esta revisión se enfoca en el diagnóstico diferencial de las causas de rabdomiólisis, haciendo un enfoque especial en aquellas de causa metabólica, y en el enfrentamiento y manejo de esta situación clínica.
FISIOPATOLOGÍAVarios mecanismos fisiopatológicos se han relacionado a la rabdomiólisis6, entre las patologías que afectan y predisponen al musculo a ella destacan:
- (1)
Metabólicas: Falla en la producción de energía: como en los defectos de la β-oxidación de ácidos grasos, mutaciones en gen de la LPIN1, errores innatos de la glicogenolisis y glicolisis, defectos de la cadena respiratoria mitocondrial, de las purinas y de la α-metil-acil-CoAracemasa (AMACR) peroxisomal.
- (2)
Estructurales: En las distrofias y miopatías
- (3)
Alteración en la bomba de calcio en las mutaciones en el gen RYR1.
- (4)
Inflamatorios en las miositis.
En el sarcolema de manera normal las bombas iónicas y canales mantienen una baja concentración intracelular de sodio y calcio y una alta concentración de potasio intracelular.
Independiente de la causa de la rabdomiólisis, la vía final común es la injuria del sarcolema, ya sea por aumento del calcio intracelular o por falla de la producción de energía por disfunción de la bomba de Na+/K+ATPasa y Ca2 +ATPasa. Altas concentraciones de sodio y calcio intracelular actúan de manera nociva por diversos mecanismos, por un lado perpetúan la contracción muscular depletando la energía celular y por otro activan proteasas y fosfolipasas calcio dependientes, que contribuyen a la destrucción de las proteínas de la membrana y del citoesqueleto, lo que conduce a la necrosis de la fibra, consecuentemente se liberan a la circulación electrolitos (principalmente potasio) y proteínas intracelulares (aldolasa, mioglobina, creatinkinasa, lactato deshidrogenasa, aspartato transaminasa)7.
La rabdomiólisis frecuentemente es precipitada por fiebre, enfermedades infecciosas, ejercicio (especialmente en condiciones de ayuno), condiciones asociadas a temperatura elevada y circulación de mediadores proinflamatorios como las citokinas y quimiokinas8.
Se presenta con debilidad y aumento de volumen doloroso de los músculos, aunque puede ser incluso asintomática, y mioglobinuria detectada clínicamente por orina de color café.
Dependiendo de la severidad pueden presentarse complicaciones como síndrome compartimental (debido al daño toxico a capilares con edema secundario del músculo), insuficiencia renal (por efecto tóxico de la mioglobina), hiperkalemia y coagulación intravascular diseminada (por liberación de tromboplastina a la circulación). Los riesgos más importantes son la insuficiencia renal, respiratoria y arritmias cardíacas9.
La tasa de mortalidad de rabdomiólisis es aproximadamente 8–10%10, la mayor mortalidad está asociada a insuficiencia renal (complicación que se presenta en un 15 a 50% de los casos) o arritmia con paro cardiaco debido a hiperkalemia11.
En la mayoría de los pacientes la CK es normal entre los episodios agudos de rabdomiólisis, excepto en casos de distrofias musculares, miositis o defectos en el metabolismo del glicógeno.
CAUSAS DE RABDOMIÓLISISEn un estudio en 191 pacientes pediátricos las causas más frecuentes correspondieron a trauma y miositis viral, constituyendo el 64% de los pacientes. En los pacientes diagnosticados como miositis viral (n=73), 4 fueron positivos para influenza B y 2 eran positivos para la influenza A12. En los casos de rabdomiólisis recurrente, o con antecedentes personales o familiares de mialgias post ejercicio, la causa más frecuente son deficiencias enzimáticas. Es importante recordar que infecciones, tóxicos o medicamentos pueden ser el gatillante de crisis metabólicas por errores innatos del metabolismo lipídico, carbohidratos o purinas.
Las anormalidades metabólicas más frecuentemente reportadas, tanto en niños como adultos, han sido la deficiencia de Carnitina Palmitoil transferasa II (CPT II), seguida de los defectos de glicólisis (déficit de fosforilasa y fosfoglicerato kinasa fosfoglucomutasa), aunque es necesario considerar una extensa lista de posibilidades3,6. Pacientes con antecedentes de intolerancia al ejercicio, mialgias, hipertermia maligna, presentan mayor riesgo de rabdomiólisis.
Recientemente se han descrito mutaciones en el gen LPIN1 que codifica para la enzima Lipina 1, como nueva causa de rabdomiólisis severa en la infancia. Las lipinas son una familia de enzimas, aún no totalmente caracterizadas, relevantes en el metabolismo de los triglicéridos. El gen LPIN1 codifica para la ácido fosfatídico fosfatasa músculo-específica, enzima clave en la biosíntesis de triglicéridos y fosfolípidos de membrana. Un estudio de 29 pacientes con rabdomiólisis mostró que 17 (59%) presentaban mutaciones en el gen LPIN1. En estos 17 pacientes, los episodios de rabdomiólisis se produjeron en una edad promedio de 21 meses, la mayoría asociados a cuadros febriles. Se ha considerado que mutaciones en este gen predispondrían a la miopatía relacionada con el uso de estatinas como agentes hipocolesterolémicos en portadores14.
En el déficit de CPT II, la mioglobinuria puede ocurrir con o sin ejercicio, con ayuno prolongado o con combinación de ayuno y ejercicio15 a diferencia de las glicogenosis en que siempre es desencadenada por ejercicio.
En las enfermedades mitocondriales, la intolerancia al ejercicio es un elemento frecuente y puede ser el síntoma único en los defectos de los complejos de la cadena respiratoria I, IV y más comúnmente del III. Se acompañan de acidosis láctica en reposo o tras ejercicio moderado y de alteraciones histoquímicas y enzimáticas en músculo que definen el diagnóstico. Se han descrito múltiples mutaciones mitocondriales asociadas a mioglobinuria ya sea aislada o como parte de un cuadro más complejo como el MELAS16.
Déficit de mioadenilato deaminasaSe manifiesta por mialgias post ejercicio, calambres, e intolerancia al ejercicio, de inicio en la infancia, adolescencia o edad adulta. Evoluciona con síntomas moderados a severos durante los primeros años, pero luego el curso clínico generalmente se estabiliza. Es un trastorno hereditario autosómico recesivo producido por mutaciones en el gen AMPD1, que afectan el ciclo de nucleótidos de purina. La mayoría de los pacientes son homocigotos y la mutación más frecuente en este gen es la C34T, presente en cerca del 1% de la población caucásica, pero la mayoría asintomática. El diagnóstico se basa en histoquímica y análisis bioquímico en músculo, o en la identificación de las mutaciones causales.
Se ha descrito déficit de la actividad enzimática de la isoforma de eritrocitos de AMP deaminasa en pacientes con bajos niveles de ácido úrico, sin signos clínicos evidentes. Se ha reportado mejoría de síntomas tras ingesta de D-ribosa, con efectos transitorios17.
OTRAS CAUSAS DE RABDOMIÓLISISLas miopatías, y especialmente las distrofias musculares se asocian a mayor riesgo de rabdomiólisis y de hipertermia maligna18. Fenotipos leves de distrofinopatías, con signos clínicos sutiles pueden presentarse como mialgias pos ejercicio y menos frecuentemente mioglobinuria; los niveles séricos de CK entre episodios son en general más altos que en las glicogenosis19.
Síndrome serotoninérgico: el exceso de actividad serotoninérgica genera alteración del estado mental, irritabilidad neuromuscular e inestabilidad autonómica debido a excesiva activación de receptores 5-HT1A y 5-HT2. Casos graves se han asociado a rabdomiólisis con insuficiencia renal20.
LABORATORIOLos estudios recomendados en rabdomiólisis se presentan en la Tabla 2.3 En la evaluación inicial deben descartarse trastornos adquiridos potencialmente tratables que pueden causare mialgias e HiperCKemia como son: la deficiencia de vitamina D, enfermedad tiroídea, trastorno metabólico o hidroelectrolítico, enfermedad intercurrente o precedente (infecciones virales, fármacos, medicamentos, tóxicos, etc)6.
Estudio de laboratorio en rabdomiólisis en niños
| Sangre | -Creatinkinasa, lactato deshidrogenasa |
| -Electrolitos, funcion renal y hepatica | |
| -Hemograma y frotis | |
| -Perfil de coagulación | |
| -Gases arteriales, glucosa, lactato, cuerpos cetónicos, amonio | |
| -Perfil de acilcarnitinas en ayuno | |
| -Carnitina total, libre y esterificada | |
| -Ácidos grasos libres | |
| -Función tiroidea | |
| -Niveles de vitamina D | |
| -Marcadores inflamatorios | |
| -Serología de Epstein-Barr y CMV | |
| -Frotis faríngeo para virus respiratorios | |
| Orina | -Ácidos orgánicos en Orina, incluidos ácidos dicarboxílicos y acilglicina |
| -Aminoácidos | |
| -Mioglobinuria | |
| LCR | -Lactato |
| Otros | -ECG |
| -Ecocardiograma | |
| -Estudios de neuroconducción | |
(Modificado de: Chan EK, Kornberg AJ, Ryan MM. A diagnostic approach to recurrent myalgia and rhabdomyolysis in children. Arch Dis Child. 2015;100(8):793–7).
La CK sérica generalmente alcanza niveles sobre 100 veces lo normal. Puede coexistir hiperuricemia, hiperfosfatemia e hipo o hipercalcemia. Si se desarrolla insuficiencia renal pueden subir los niveles de potasio y calcio. La orina es positiva para mioglobinuria, hemoglobinuria sin hematuria y negativa para porfiria.
La biopsia muscular muestra necrosis y degeneración de fibras musculares, inflamación o alteraciones estructurales7.
Según la magnitud de la elevación de la CK y la edad del paciente la aproximación al diagnóstico y diagnóstico diferencial se muestra en la Figura 1.21
MEDIDAS DE MANEJO EN RABDOMIÓLISIS:793–7)")
En la fase aguda el tratamiento tiene como objetivo preservar la función renal y restaurar anormalidades metabólicas. Se inicia con aporte precoz y adecuado de volumen con NaCL 0.9% ev. (sin potasio o lactato)9. Se recomienda comenzar a un ritmo de 1.5 lt/h, en niños 10 a 15ml/kg, manteniendo una diuresis de 200–300ml/h, hasta que la creatinkinasa se reduzca a menos de 1000 UI/l, manteniendo monitorización de función cardiaca y pulmonar por efectos secundarios de hipervolemia. Aún cuando no hay evidencia adecuada, se recomienda agregar manitol (después de la infusión de volumen y en pacientes sin oliguria) y bicarbonato. La alcalinización de la orina (pH 6.5) ayuda a aclaramiento de mioglobina y a corregir la acidosis metabólica y por lo tanto la hiperkalemia. Debe considerarse, en situaciones de acidosis o hiperkalemia extremas, la hemodiálisis diaria o terapias de reemplazo renal continuas con membranas hiper permeables que permitan remover además la mioglobina de alto peso molecular de la circulación22. Complicaciones importantes son la coagulación intravascular diseminada y síndromes compartimentales que pueden requerir fasciotomías múltiples precoces.
En mutaciones en gen de la Lipina 1 (LPIN1) que llevan a déficits de energía en la fibra muscular, causantes de hasta el 50% de los episodios de rabdomiólisis en pacientes pediátricos, se han propuesto estrategias terapéuticas dirigidas a la prevención y tratamiento precoz del catabolismo, que incluyen el aporte de soluciones intravenosas con elevadas concentraciones de glucosa, en orden a asegurar un óptimo aporte calórico. En una serie pequeña de pacientes, esta intervención redujo la duración del episodio de 7-10 días a 523.
Medidas recomendadas en casos de rabdomiólisis en mutaciones del gen de la LPIN1:
- 1
Hospitalización inmediata con acceso a unidad de pacientes críticos, por riesgos de arritmias asociadas a hiperkalemia e insuficiencia renal, incluso si niveles de CPK son normales (monitorización por riesgo de elevación de CK más tardía). La hospitalización debe mantenerse hasta al menos 24 horas tras normalización de CK o tras 48 horas en caso de dolor muscular persistente.
- 2
Monitorización hemodinámica, respiratoria y del balance hidroelectrolítico (monitorización de diuresis cada 3 horas) durante la hospitalización.
- 3
Proveer de 2 accesos venosos periféricos o centrales, que permitan hiperhidratación y toma de muestras fácil sin inducir hemólisis (kalemia confiable).
- 4
Mantener aporte nutricional en base a carbohidratos, evitando lípidos.
- 5
Considerar aporte de esteroides en ausencia de contraindicación de uso: dexametasona o metilprednisolona 2mg/kg/día por 2-5 días eu12.
- 6
Hiperhidratación: 3litros/m2/día [superficie corporal=(4×P+7)/(P+90)] sin esperar aumento de kalemia o CPK. Verificar aporte por litro de: 200ml de glucosa 30% (180gramos/m2 equivalente a 6gr/Kg/24h)+400ml de bicarbonato isotónico 14%+400ml de fluido salino 0.9%, sin adición de potasio ni calcio.
- 7
Considerar aporte de carnitina 100mg/kg al día.
- 1.
Mediciones seriadas de electrolitos en sangre (Na, K, Ca, pH sérico, Mg), glicemia, urea y creatinina; CKy o buscar mioglobinuria.
- 2.
Si tiene fiebre: búsqueda de foco infeccioso, incluyendo hemocultivos.
- 3.
ECG y Ecocardiografía para buscar compromiso del miocardio y evaluar función ventricular izquierda y la tolerancia a la hiperhidratación.
- 4.
Mediciones de sodio y potasio cada 2 horas durante las primeras 24 horas+nivel de glucosa capilar. Luego mediciones de electrolitos en sangre cada 6 horas.
- 5.
Estimación de diuresis horaria >2ml/kg/hora (catéter urinario).
- 6.
Monitoreo del pH urinario y la densidad urinaria ≤1005.
- 7.
Evaluar el balance hídrico cada 3horas.
Las autoras no tienen ningún conflicto de interés que declarar.
Referencia no citada