







Se han producido recientemente importantes avances terapéuticos en la atrofia muscular espinal (AME) por alteración del gen SMN1, conocida también como AME 5q por la localización cromosómica de dicho gen. Descrita en el siglo XIX e identificado el gen causante en 1995, durante estas últimas décadas el esfuerzo colaborativo de investigadores, profesionales sanitarios y grupos de familias ha ayudado a aumentar los conocimientos de la genética de la enfermedad, clave para el desarrollo de terapias avanzadas. Destaca la investigación clínica y aprobación de la terapia antisentido con Nusinersen como primer tratamiento específico para la enfermedad y otras, como la terapia génica, con resultados muy prometedores en pacientes que, por primera vez, hacen que la AME deje de considerarse una enfermedad intratable. En el camino hacia una posible cura o cambio radical en el curso clínico, cobran mayor importancia el diagnóstico precoz, la implementación de registros, el seguimiento multidisciplinario y la uniformidad de cuidados en diferentes especialidades para optimizar al máximo la eficacia de los tratamientos. La AME constituye un claro ejemplo para otras enfermedades neuromusculares raras de origen genético demostrando que la investigación de las causas genéticas puede ayudar a descubrir y desarrollar terapias efectivas y con importantes beneficios para los pacientes y sus familias.
Recently, there have been important therapeutic advances in spinal muscular atrophy (SMA) caused by alteration of the SMN1 gene, also known as SMA 5q due to its chromosomal location. Described in the XIX century and identified the causative gene in 1995, during these last decades the collaborative effort of researchers, health professionals and advocacy groups contributed to increase the knowledge of the genetics of the disease, key to develop advanced therapies. Results on clinical research and approval of antisense therapy with Nusinersen as the first specific treatment for the disease as well as gene therapy, with very promising results in patients, make no longer SMA being considered as an intractable disease. On the way to a possible cure or substantial change in the clinical course, it is important to support early diagnosis, the implementation of registries, multidisciplinary follow-up and the uniformity of standard of care in different specialties to optimize the efficacy of treatments. SMA is a clear example for other rare neuromuscular diseases of genetic origin demonstrating that the investigation of genetic causes can help to discover and develop effective therapies with important benefits for patients and their families.información del artículo
La Atrofia Muscular Espinal (AME) es una enfermedad con un patrón de herencia autosómico recesivo que cursa con degeneración y pérdida de las neuronas motoras de la médula espinal, produciéndose denervación y debilidad muscular. La AME fue descrita originalmente por Guido Werning y Johann Hoffmann entre 1891 y 19001, y un siglo después, los avances genéticos permitieron la localización e identificación del gen SMN1 como causante de la enfermedad2.
La AME se considera una de las enfermedades raras y neuromusculares más frecuentes y que ocasiona una importante morbimortalidad en la edad infantil con consecuencias perjudiciales para el paciente, la familia y el sistema de salud3. Afecta a una de cada 6000-10000 recién nacidos y la frecuencia de portadores es de 1 cada 40 o 50 individuos de la población general. La AME consiste en un continuo de manifestaciones clínicas de debilidad muscular y denervación que van desde formas graves congénitas hasta manifestaciones mínimas en la edad adulta4. A pesar de ser un continuo, se ha clasificado en tres grandes tipos según la edad de inicio de los síntomas y la función motora máxima alcanzada con distintos subtipos (Tabla 1)4. La forma más grave Tipo 1 se manifiesta en las primeras semanas o meses de la vida y son pacientes con hipotonía generalizada, difícilmente llegan a tener sostén cefálico y nunca llegan a sentarse. La historia natural predice que, si no se realiza una intervención que implique llegar a la traqueostomía y la ventilación mecánica invasiva, más de la mitad habrán fallecido por problemas respiratorios durante el primer año y casi todos los casos al segundo año de vida5,6. Existe una forma de fenotipo extremadamente grave, congénita con contracturas, cardiopatía y necrosis vascular que fallece a los pocos días o semanas, (conocida AME tipo 0 o tipo 1A)7. Cuando el comienzo ocurre después de los 6 meses, los pacientes ya adquieren la capacidad de sedestación, pero no logran llegar a la deambulación espontánea estando confinados permanentemente a la silla de ruedas. La mayoría llega a la edad adulta aunque con complicaciones respiratorias, escoliosis y contracturas. Siguiendo la clasificación, son conocidas como formas tipo 2 y hay pacientes débiles que nunca llegan a ponerse de pie y otros menos comprometidos que con ayuda pueden mantenerse y dar algunos pasos, pero nunca llega a ser una deambulación independiente. Si la debilidad muscular aparece después que el paciente deambula, estamos frente a pacientes con la forma tipo 3, que pierden esa capacidad luego, en los años siguientes. También existen casos menos afectados que mantienen la deambulación autónoma por muchos años e inclusive pueden manifestar la enfermedad en la segunda o tercera década de la vida o posterior (más detalles en la Tabla 1).
Clasificación considerando un espectro continuo de la AME
| Tipo AME | Edad Inicio | Función motora lograda | Evolución, historia natural, complicaciones | Copias SMN2 representativas | Comentarios |
|---|---|---|---|---|---|
| 1A (también referido como tipo 0) | Prenatal/Congénita | Ninguna | Fallecimiento en pocas semanas, cardiopatía, contracturas, trastornos vasculares | 1 | Algunos autores consideran la tipo 0 diferente a la tipo 1A incluyendo en esta última formas de inicio en pocos días de vida aunque en estos casos, los pacientes tendrían (como los tipos 1B) dos copias de SMN2 y sin fenotipo extremo de cardiopatía, contracturas, trastornos vasculares |
| 1B | <3M | Control cefálico parcial en algunos pacientes | Problemas respiratorios y para alimentarse Declinación lineal Fallecimiento en dos o tres años sin intervención invasiva (traqueostomía, respirador) | 2 | Pueden manifestarse a los pocos días o semanas de vida (algunos autores indican que estos casos tan precoces pueden ser tipo 1A) |
| 1C | >3M | Control cefálico | Problemas respiratorios y para alimentarse Pueden mantener la poca función motora alcanzada | 3 | En general más estables que la forma tipo 1B pero, pueden tener mismas complicaciones |
| 2A | >6M | Se sientan pero no llegan a ponerse de pie | Escoliosis Llegan a la adolescencia y vida adulta, pero con complicaciones Períodos de poca estabilidad | 3 | Algunos casos pueden empezar antes de los 6 meses. En general se consideran tipo 2 débiles y algunos pueden inclusive perder la capacidad de sentarse en el futuro |
| 2B | >12M | Se sientan y llegan a alcanzar la bipedestación | Escoliosis Llegan a la adolescencia y vida adulta con menos complicaciones Períodos de estabilidad | 3 | Algunos casos pueden empezar antes del año. En general se consideran tipo 2 fuertes y pueden inclusive llegar a dar algunos pasos con ayuda |
| 3A | Entre los 18 y los 36 meses | Llegan a caminar sin ayuda | Escoliosis Pierden la deambulación a una edad temprana Períodos de estabilidad | 3 | La pérdida de deambulación se asocia a algunas complicaciones que se ven pacientes tipo 2 |
| 3B | >3 años | Caminan sin ayuda | Pierden la deambulación más tarde Más estables que los tipo 3A | 3-4 | Algunos pacientes llegan a deambular siempre o hasta edades avanzadas |
| 4 | Segunda o tercera década de la vida | Caminan sin ayuda | Suelen deambular toda la vida sin mayores complicaciones Muy estables | 4 o más | Pueden ser hermanos/as menos graves de pacientes tipo 3 |
| 5 (Manifestaciones mínimas o asintomáticos) | No suelen detectarse | Pautas motoras normales | En general se los considera asintomáticos aunque pueden tener calambres o hallazgos electromiográficos | 4 o más | Suelen ser hallazgos moleculares positivos (SMN1 -/-) en hermanos/as de pacientes tipo 3/4 |
Clasificación considerando un espectro continuo de la AME de acuerdo con la edad de comienzo, función motora, evolución, complicaciones y número de copias de SMN2 (basado en referencias 4,5,7,24,30). La AME tipo 5 es posible considerarla cuando una persona tiene genéticamente confirmado AME por alteración de SMN1 pero no manifiesta la enfermedad o es virtualmente asintomática.
La función que tienen los genes es llevar la información del ADN para que se sinteticen las proteínas del organismo, a través del ARN mensajero (mRNA). Este proceso de síntesis de proteínas se realiza en dos etapas: la transcripción, que consiste en la síntesis de mRNA a partir de ADN y la traducción que consiste en la síntesis de proteínas por los ribosomas a partir del mRNA.
La copia de una de las dos cadenas del ADN da como resultado el transcrito primario o pre-mRNA. Este transcrito primario tiene regiones que hay que eliminar, además de añadirle unos suplementos para que se estabilice. Estas regiones a eliminar, llamadas intrones, se llevan a cabo por un proceso molecular denominado splicing. En este proceso se eliminan los intrones y lo que queda del pre-mRNA (los exones), se empalman unos a otros formándose, ahora sí, el ARN mensajero maduro que sale del núcleo al citoplasma para la síntesis proteica.
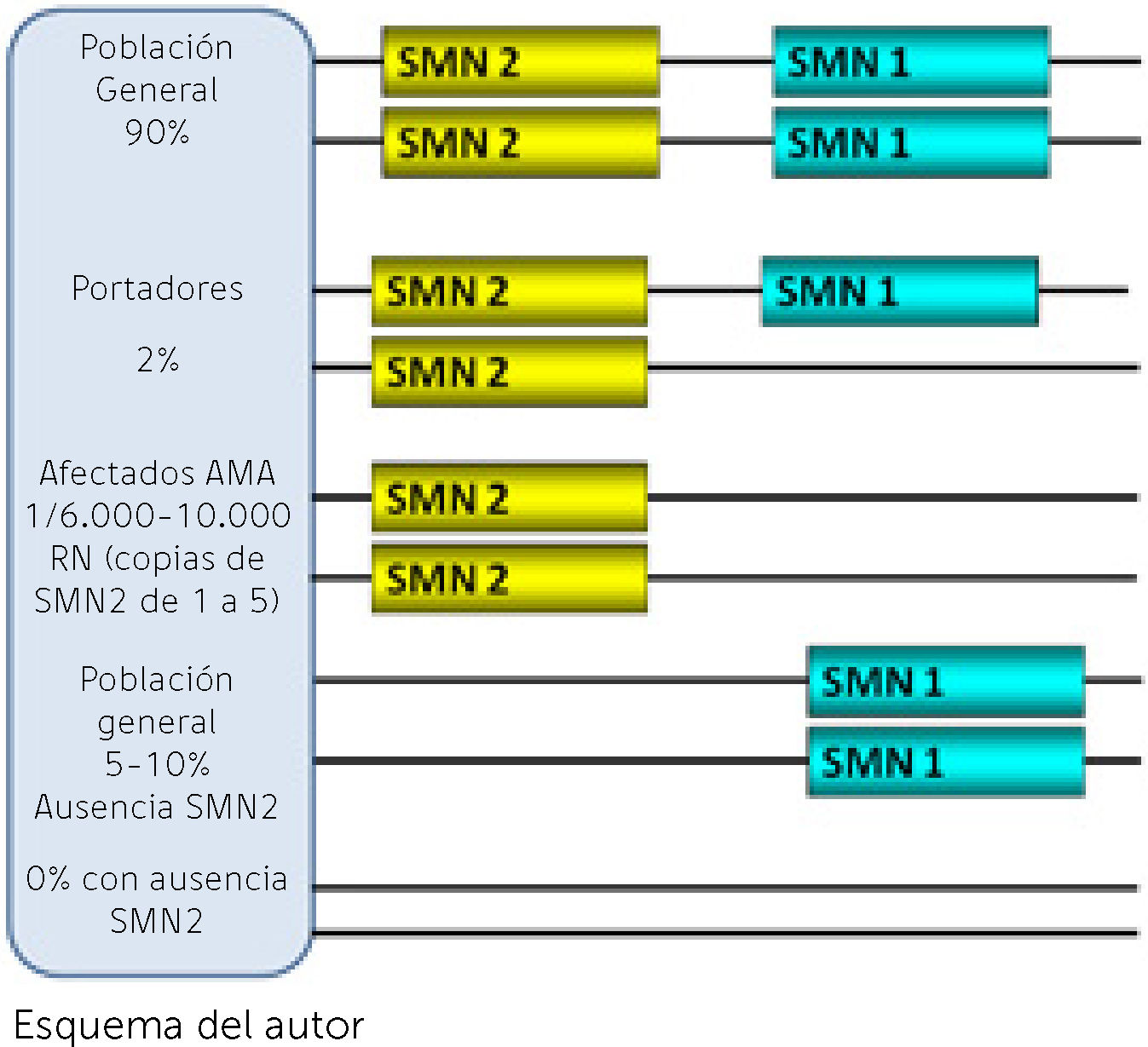
La región que involucra a los genes de la AME en el brazo largo del cromosoma 5 está duplicada habiendo entonces dos versiones de varios genes, caracterizadas de manera general como telomérica y centromérica. La AME involucra a dos de esos genes, SMN1 (telomérico) ya mencionado, como el determinante de la enfermedad dado que su ausencia o mutaciones constituyen la confirmación genética de la sospecha clínica. El otro gen, SMN2 (centromérico), tiene una altísima homología con SMN1 y se considera como modificador del fenotipo (Figura 1). El 95% de los pacientes carecen de SMN1 y el resto pueden tener mutaciones puntuales8. En cambio los pacientes siempre tienen de 1 a 5 copias del gen homólogo SMN2 que produce una proteína en su mayoría incompleta por ausencia del exón 79. La exclusión del exón 7 del pre-mRNA y por ende del RNA mensajero y la proteína SMN ocurre por una transición citosina (C) a timina (T) en dicho exón considerada una mutación silenciosa dado que no predice un cambio de aminoácido pero, que tiene efectos perjudiciales dado que facilita la unión de dicho exón con proteínas inhibidoras para que lo excluyan del proceso de splicing10 (Ver Figura 2 A y B). Es así que, a diferencia del SMN1, que es en altísimo porcentaje completa, la gran mayoría de proteína que produce SMN2 es incompleta sin la parte del exón 7 (delta 7) que la hace parcialmente funcionante y rápidamente degradable. Todas las formas clínicas de AME tienen ausencia de SMN1 y el número de copias de SMN2 se correlaciona con el fenotipo. Los pacientes con la forma más grave tipo 1 tienen en su gran mayoría 2 copias de SMN2. Los pacientes tipo 2 tienen mayoritariamente 3 copias y los pacientes tipo 3 entre 3 y 4 copias. La correlación con la clínica no es absoluta dado que existen discordancias fenotípicas (Tabla 2)9. Esta no es la única singularidad en la genética de la AME ya que solamente los humanos parecen tener la región duplicada con ambos genes SMN1 y SMN2 mientras que en el resto de especies hay una sola versión denominada Smn11. La función de la proteína SMN es fundamental en todas las células del organismo y su ausencia total tiene efecto letal in utero. Es por eso que no existe un modelo natural animal de la enfermedad y tampoco se ha descrito ningún ser humano con ausencia de ambos genes SMN (Figura 1). La menor cantidad de proteína completa producida por el gen SMN2 es suficiente para poder nacer y desarrollarse aunque no evita la aparición de la enfermedad. El conocimiento de estas bases genéticas de la enfermedad fue fundamental para lograr la generación a partir del año 2000 de modelos de ratones transgénicos, knock-down y condicionados que mimetizaran la enfermedad y en los que probar alternativas terapéuticas12. Hace más de 20 años que se conoce el gen y la proteína y existen varias funciones que se han demostrado in vitro e in vivo relacionadas con el metabolismo y el splicing en todas las células incluyendo el transporte axonal de RNA13. Pero la función específica de SMN en las neuronas motoras y sobre todo lo que las hace a éstas más sensibles a la disminución de SMN es todavía cuestión de debate14.

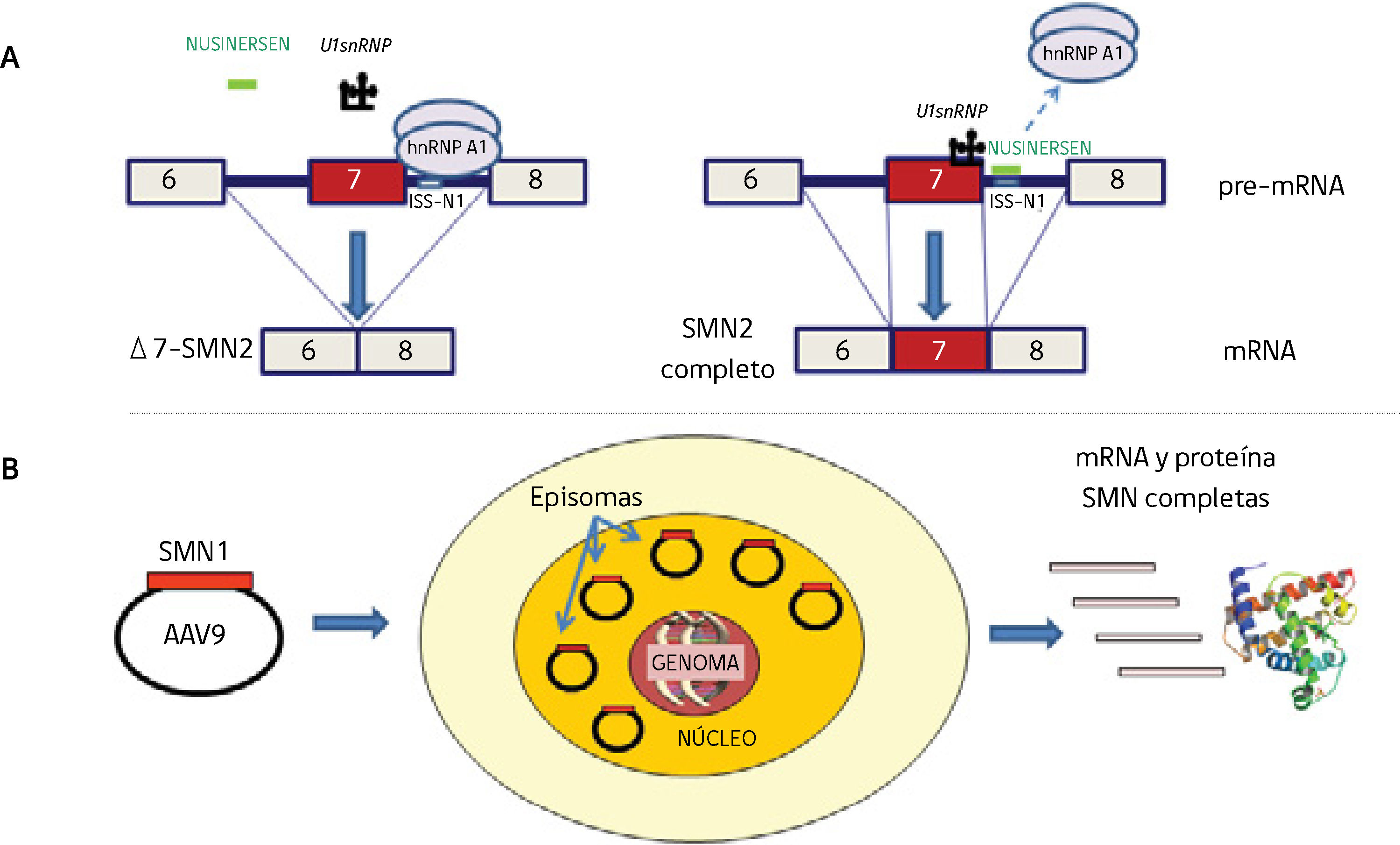
 Mecanismo de acción de la terapia antisentido con Nusinersen. El ISS-1 (Intronic splicing silencer 1) ubicado en el intrón 7 del gen SMN2 atrae la proteína hnRNP A1 que impide que el U1snRNP reconozca la parte final del exón 7 para que se una al exón 8. En presencia de Nusinersen (oligonucleótido antisentido de 18 bases) que se une al ISS-1, la proteína hnRNP A1 no se une y deja sitio para que U1snRNP reconozca el exón 7 (basado en referencias 4,20-22). B) Terapia génica. La construcción self complementaryAAV9 y el cDNA del gen SMN1 llegan al núcleo, no se integran en el genoma y se comportan como episomas con replicación propia independiente que produce RNA y proteína SMN completas. Si la célula se divide, estos episomas se repartirían entre las células hijas, lo que hace efectiva esta terapia en células que no se dividan, como ocurre en las neuronas motoras (basado en referencias 27,30).")
Terapias avanzadas en la atrofia muscular espinal
A) Mecanismo de acción de la terapia antisentido con Nusinersen. El ISS-1 (Intronic splicing silencer 1) ubicado en el intrón 7 del gen SMN2 atrae la proteína hnRNP A1 que impide que el U1snRNP reconozca la parte final del exón 7 para que se una al exón 8. En presencia de Nusinersen (oligonucleótido antisentido de 18 bases) que se une al ISS-1, la proteína hnRNP A1 no se une y deja sitio para que U1snRNP reconozca el exón 7 (basado en referencias 4,20-22).
B) Terapia génica. La construcción self complementaryAAV9 y el cDNA del gen SMN1 llegan al núcleo, no se integran en el genoma y se comportan como episomas con replicación propia independiente que produce RNA y proteína SMN completas. Si la célula se divide, estos episomas se repartirían entre las células hijas, lo que hace efectiva esta terapia en células que no se dividan, como ocurre en las neuronas motoras (basado en referencias 27,30).
Discordancias fenotípicas en AME
| Fenotipo menos grave que el esperado |
| - Pacientes con dos copias de SMN2 pero, que evolucionan como tipo II o III: Portadores variante c.859G>C en SMN2 y otros modificadores |
| Fenotipo más grave que el esperado |
| -Pacientes con tres copias de SMN2, pero, que evolucionan como tipo I: Metilación, deleción parcial o variante de alguna de las copias de SMN2 |
| -Pacientes con cuatro copias de SMN2 pero, que evolucionan como tipo II: Metilación, deleción parcial o variante de alguna de las copias de SMN2 |
| Hermanos/as de pacientes con ausencia o mutación en homocigosisSMN1y que tienen manifestaciones mínimas o asintomáticos |
| -No equivalencia de las copias de SMN2 |
| -Influencia de otros genes o factores modificadores |
(Basados en referencia 9).
Ante la sospecha clínica de un paciente con AME, el primer paso diagnóstico lo constituye el estudio genético, que establecerá la ausencia o presencia de SMN1. A partir de la confirmación genética, se inicia el proceso de comunicación a la familia, donde hay que considerar distintos aspectos según la edad de comienzo de las manifestaciones, la edad del paciente y la función motora máxima alcanzada, que nos establecerán en la mayoría de los casos el tipo de AME de dicho paciente. Considerando el número de copias de SMN2, si bien la correlación para predecir el tipo de AME es importante9, no llega a ser absoluta por las discordancias conocidas (Tabla 2).
En los casos más graves, hasta hace poco se solía informar a la familia sobre la evolución y la historia natural, con alta probabilidad de desenlace fatal y para establecer un consenso sobre la conducta de seguimiento y la alternativa de cuidados paliativos o de intervención con traqueotomía y ventilación mecánica invasiva. El dilema ético que esto presentaba se ha ido transformando a lo largo de los resultados en este último tiempo desde la aprobación de medicación que muestra ser efectiva y específica de la enfermedad (ver apartado siguiente).
Actualmente, las medidas proactivas en vez de reactivas, forman parte de la uniformidad de los criterios de seguimiento y cuidados consensuados internacionalmente (Tablas 3 y 4)15,16.
Aspectos consensuados internacionalmente de diagnóstico, seguimiento y cuidados en los pacientes con AME
| Aspectos diagnósticos genéticos |
| Nutrición, crecimiento y cuidados óseos |
| Aspectos respiratorios |
| Cuidados ortopédicos |
| Fisioterapia y rehabilitación |
| Compromiso de otros órganos o sistemas |
| Cuidados hospitalarios del paciente agudo y las complicaciones |
| Medicaciones y tratamientos |
| Aspectos éticos y cuidados paliativos |
(Basados en ref. 15,16,30)
Consideraciones sobre el nuevo escenario terapéutico y protocolos en AME
| CONTEXTO |
| -Medidas proactivas versus reactivas |
| -Seguimiento multidisciplinar y gestión de casos |
| -Uniformidad de normas y protocolos de seguimiento |
| -Contexto biopsicosocial del paciente y su familia |
| -Manejo de las expectativas |
| ESTRATEGIAS |
| -Selección de pacientes según prioridad (gravedad, edad, evolución, expectativas) |
| -Tiempo de administración (corto-mediano-largo plazo) |
| -Eficacia terapéutica versus la realidad biológica de la enfermedad en cada paciente (pacientes respondedores o no claramente respondedores) |
| -Terapia sistema nervioso central versus periférica (músculo, unión neuromuscular, otros órganos) |
| -Combinación de terapias (diferente mecanismo acción, diferente vía y tiempo administración) |
| -Ventana terapéutica (diferente según el tipo de AME) |
| -Rescate del fenotipo o intervención presintomática (pacientes sintomáticos o presintomáticos por cribado neonatal) |
| EFECTOS |
| -Consecuencias epidemiológicas (incidencia, prevalencia) |
| -Curación, detener el proceso, cambiar de tipo de AME |
| -Manifestaciones de la enfermedad con las nuevas terapias, fenotipos emergentes, ¿nuevas clasificaciones? |
(Basados en referencias 4 y 28, más explicación texto).
Es por ello que hoy se considera esencial el seguimiento integral por un equipo multidisciplinar dado que se hace cada vez más necesario organizar y coordinar el seguimiento médico del paciente entre varios especialistas. Además de las medidas de seguimiento establecidas, es importante considerar otros aspectos, como la formalización de registros con datos de evolución de los pacientes, la toma de decisiones consensuadas con respecto a todos los aspectos del tratamiento (incluyendo la posibilidad de cuidados paliativos) y la transición entre la pediatría y la medicina de adultos. Para este fin, la actividad de los distintos especialistas se involucra cada vez más en áreas transversales con otros facultativos de las áreas asistenciales a través de la gestión de casos y contando con el entorno psicosocial del paciente (Tabla 4).
TRATAMIENTOS AVANZADOSEn el campo terapéutico, los conocimientos generados de las bases moleculares de la AME ha dado lugar a la investigación clínica de terapias avanzadas muy específicas, ya sea en la modificación o reemplazo del gen SMN1 o en la modulación del ARN (Figura 2). Publicaciones recientes en el New England Journal of Medicine describen la eficacia observada en ensayos clínicos de terapias avanzadas en pacientes con atrofia muscular espinal. Se refieren a:
- 1)
A la administración intratecal de un oligonucleótido antisentido de 18 pares de bases en dosis iniciales de impregnación con mantenimiento cada 4 meses que actúa sobre el gen SMN2, homólogo del SMN1 y que está presente en todos los pacientes17,18.
- 2)
La administración en una única inyección intravenosa de un vector viral selfcomplementary Adeno Associated Virus 9 (scAAV9) que contiene la versión normal del gen SMN119.
El Dr. Adrian Krainer y su equipo de Cold Spring Harbour en Nueva York investigaron el efecto de distintos oligonucleótidos en el mecanismo de splicing del exón 7 del SMN2. Se observó eficacia in vitro aunque para su aplicación subcutánea o intravenosa no eran capaces de pasar la barrera hematoencefálica. Realizaron entonces la inyección por vía intratecal en modelos de ratón AME con expectativa de vida de 14 días y observaron que uno de ellos que se unía específicamente a un sitio del intrón 7 (ISS-1), incluía mayoritariamente el exón 7 en el RNA e incrementaba la cantidad de proteína SMN lo que conducía a aumentar la sobrevida y mejorar las manifestaciones de debilidad muscular del ratón20,21. Con algunas evidencias más preclínicas acumuladas22, el ensayo en humanos no se hizo esperar. Patrocinado por el laboratorio farmacéutico IONIS, el oligonucleótido elegido se bautizó Nusinersen y estudios piloto previos indicaron que la administración intratecal en pacientes era segura y bien tolerada con datos preliminares de efectividad mejorando la debilidad muscular23. En 2014 se iniciaron los primeros reclutamientos de pacientes para hacer un ensayo fase 3 multicéntrico en Norteamérica, Europa y Asia que se denominó ENDEAR. Se eligió una población con pacientes afectados de AME 1 que tuvieran menos de 7 meses y dos copias de SMN2 y se reclutaron 120 pacientes hasta abril de 2016. Dos tercios de los pacientes se agrupaban al azar para recibir la medicación periódicamente cada 4 meses después de una fase de impregnación inicial y en el tercio restante se hacía un procedimiento simulado (placebo). La finalidad del ensayo era comparar la evolución de los dos grupos en relación a morbimortalidad y si mejoraban las pautas motoras medidas con escalas validadas para ese fin como Hammersmith Infant Neurological Examination sección 2 (HINE-2) y Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP intend).
La diferencia que se notó en el grupo de los pacientes AME tipo 1 con medicación (que en su historia natural no presentan control cefálico ni sedestación ni prácticamente movimiento de los miembros) fue que adquirían estas pautas motoras nunca observadas en estos pacientes24. El compromiso respiratorio y la sobrevida del grupo con medicación también fueron más favorables en los pacientes bajo tratamiento17.
Otro ensayo con la misma medicación y un diseño similar en pacientes con formas tipo II no deambulantes con edades comprendidas entre los 2 y 12 años llamado CHERISH también demostró que los pacientes mejoraban en 4-5 puntos las escalas de función motora utilizadas como Hammersmith Functional Motor Scale Expanded (HFSME) y Revised upper limb module (RULM)18. Con todos estos datos, todos los pacientes se pasaron al brazo de medicación y Nusinersen fue aprobado para todos los tipos de AME y edad por la Food Drug Administration (FDA) en diciembre de 2016 y por la European Medicines Agency (EMA) en junio de 2017. Existen actualmente más de 4000 pacientes tratados con Nusinersen en todo el mundo incluyendo los que continúan en la fase abierta del ensayo, llamado SHINE, los casos de acceso expandido y los casos con medicación recetada y autorizada.
¿Qué podemos aprender de estos ensayos? Lo primero es que, cuanto antes se administra la medicación el grado de respuesta es mayor. Así también se documenta en el ensayo NURTURE, todavía activo, que administra Nusinersen a unos 20 pacientes asintomáticos a partir de las pocas semanas de vida y que tienen una evolución casi normal25. Lo segundo, es que en algunos pacientes más afectados y frágiles con inicio de manifestaciones durante el primer mes de vida, la respuesta a Nusinersen puede ser más lenta y que las complicaciones respiratorias pueden llevar el paciente al fallecimiento (como ocurrió con algunos pacientes del ensayo), pero por causas relacionadas no con la medicación, sino con complicaciones respiratorias de la enfermedad. En ese sentido, la medicación se muestra hasta ahora bastante segura y los efectos adversos registrados hasta ahora, si bien faltan datos a largo plazo, tienen que ver fundamentalmente con la administración intratecal17. Lo tercero es que existen algunos pacientes que no incrementan la puntuación de base en las escalas funcionales17,18. Estos pacientes, si bien aparentemente no responderían, tampoco parecen declinar en su función sino que más bien se mantienen, y pueden responder luego a medida que pasa el tiempo de tratamiento. Es importante recalcar en este punto que la estabilidad de la enfermedad, aunque no hubiera progresos motores importantes, es valorada por los pacientes y familias como muy importante a la hora de decidir recibir un tratamiento26. El efecto a largo plazo de la medicación, especialmente en población adulta todavía es desconocido. Por último, es claro que para lograr una mejor eficacia de la medicación se deben siempre aplicar las medidas de seguimiento y cuidados integrales en el contexto vigente consensuado (Tabla 3) como el sostén de la parte respiratoria con ventilación no invasiva, el soporte nutricional y una adecuada rehabilitación y fisioterapia15,16.
Terapia GénicaEn el estudio de terapia génica el equipo del Dr. Brian Kaspar y Arthur Burghes de la Universidad de Ohio demostraron que el scAAV9 era eficaz para pasar la barrera hematoencefálica y llegar a las neuronas motoras. Además no se insertaba en el genoma sino que se mantenía en el núcleo como episoma transcribiendo el gen SMN1 que lleva como inserto (Figura 2). La administración precoz de una sola inyección intravenosa en el ratón AME indicaron una altísima sobrevida, función motora conservada y tropismo neuronal27. Uno de los aspectos a considerar era que existía el efecto no solo central (en la médula espinal) sino también periférico como en la unión neuromuscular, músculo y otros órganos que pueden ser diana de la enfermedad28. Otro aspecto interesante es que al ser self complementary, la replicación y producción de RNA y proteína se haría a partir de pocas horas de introducirse en la célula, lo que facilitaría un efecto inmediato. La FDA aprobó el ensayo que patrocinó AVEXIS en pacientes AME tipo I menores de 6 meses con ausencia de SMN1 y dos copias de SMN2. El primer paciente recibió la terapia en mayo de 2013 con una dosis de 6.7 X1013 genomas virales (gv)/kg de peso. A partir del cuarto paciente reclutado la dosis se incrementó a 2.0 X1014 gv/kg, que constituye la mayor inyectada hasta la fecha en humanos19. En total se incluyeron 15 pacientes y los datos de corte en agosto de 2017 publicados indica que todos ellos habían superado los 20 meses de edad sin efectos adversos relacionados con la terapia salvo una elevación de las enzimas hepáticas transitoria que se previno con la administración de corticoides. Todos los pacientes del ensayo incrementaron la función motora, y del grupo que recibió la dosis más alta, 10 han logrado la sedestación y dos deambulan19. Actualmente se están empezando ensayos clínicos en pacientes sintomáticos y presintomáticos en EE.UU. y Europa para incrementar el número de pacientes y confirmar los datos del primer estudio (www.clinicaltrials.gov). Existen todavía algunos interrogantes con respecto al tiempo de duración del efecto de la terapia génica, aunque estudios previos con primates indican que el vector en las células podría estar varios años produciendo RNA y proteína. Por el momento no se plantea dar una repetición de la dosis dado las reacciones inmunológicas a la cápside viral que podrían producirse. En pacientes más grandes con peso mayor de 8kg, la carga viral y la biodistribución podría ser significativa y se ha protocolizado su inyección intratecal en vez de intravenosa.
Asimismo se están llevando a cabo en la atrofia muscular espinal otros ensayos clínicos y terapias experimentales incluyendo medicamentos por vía oral que modifican la inclusión del exón 7 (aunque con un mecanismo de acción diferente a Nusinersen) y con resultados que también son prometedores29 y otras terapias independientes del gen SMN1 y SMN2 como los neuroprotectores, estabilizadores de la unión neuromuscular y activadores de la función muscular (www.clinicaltrials.gov).
CONCLUSIONES Y PERSPECTIVASLa suma de todas las investigaciones clínicas y el mayor conocimiento de la evolución de la enfermedad implica distintas consideraciones relacionados en la terapia, incluyendo el contexto biopsicosocial de tratamiento, las distintas estrategias y los posibles efectos (enumerados en la Tabla 4). El mensaje es, que hay que integrar el tratamiento de la AME como parte de un engranaje que implica el seguimiento uniforme y consensuado de los pacientes15,16. De nada valdría indicar un tratamiento sin seguir estos protocolos de seguimiento. Es evidente que si se quiere iniciar una terapia precoz, los pacientes prioritarios para el tratamiento son los de diagnóstico reciente para evitar el mayor compromiso de neuronas motoras. Sin embargo, también hay que evaluar los resultados a largo plazo en pacientes históricos, que llevan mucho tiempo con la enfermedad para establecer los posibles beneficios. El seguimiento de pacientes adultos es una asignatura pendiente que habrá que establecer con protocolos específicos. En un futuro próximo será posible establecer protocolos de tratamiento combinados (distintos mecanismos de acción, central y periférica, dependiente o independiente SMN) para lograr una mayor eficacia aunque el gran desafío para los equipos de seguimiento multidisciplinar lo constituye la evolución de los pacientes tratados donde se esperan cambios clínicos y fenotipos emergentes que habrá que considerar en el contexto de cada paciente30. La alta incidencia de la AME y el beneficio de una intervención temprana hacen que pueda considerarse dentro de las enfermedades a incluir en los programas de cribado neonatal dado que con una prueba genética sencilla puede confirmarse en pocos días en el 95% de los casos por lo que el escenario de tratamiento se desviaría esencialmente a los casos presintomáticos. Los falsos negativos del cribado serían aquellos casos minoritarios con mutación puntual y también es parte del debate ético si se debería realizar la detección de portadores en el mismo cribado neonatal. También puede considerarse el cribado de portadores de una copia de SMN1 en la población general31,32 aunque alrededor del 4-5% de los portadores pueden tener dos copias de SMN133. Es evidente que se debe realizar una acción conjunta sanitaria que involucre a todos los sectores para lograr un diagnóstico más precoz, y difundir y dar a conocer las características de la enfermedad. Es esencial realizar una buena comunicación del diagnóstico, asesoramiento genético y consensuar la toma de decisiones con la familia con respecto a realizar tratamiento, elegir medidas proactivas o, en su defecto la posibilidad de medidas paliativas sin realizar tratamiento específico.
La AME constituye un ejemplo positivo para otras enfermedades raras que se están investigando no solo neuromusculares, sino en las que se conocen las causas genéticas. Se demuestra que la investigación para llegar a terapias efectivas es posible y con importantes beneficios para los pacientes y sus familias. Aún así, los altos costes de este tipo de terapia implica un compromiso tanto de las autoridades sanitarias como de la industria farmacéutica para garantizar la equidad y acceso a dichos tratamientos a toda la población de pacientes34. Los pacientes AME suelen tener una alta capacidad intelectual que los hace totalmente integrados a la sociedad si pueden mejorar sus limitaciones motoras35.
Todavía no está claro porqué las neuronas motoras son más sensibles a la disminución de la proteína SMN y se debe seguir investigando. Sin embargo, esto no ha sido un escollo para poner en claro otros aspectos de la enfermedad como la modificación del mecanismo básico genético del SMN1 y SMN2 y gracias a la investigación, lograr que la AME deje de considerarse a partir de ahora, una enfermedad sin tratamiento.
Declaración de interesesFinanciaciónEste trabajo fue parcialmente financiado por una Beca de la Fundación Privada Daniel Bravo Andreu. EFT recibió apoyo financiero para llevar a cabo ensayos clínicos en AME de Ionis/Biogen y es consultor de Biogen, AveXis, Roche.