




La Enfermedades Neuromusculares (ENM) son un conjunto de enfermedades con síntomas clínicos que varían según la edad de presentación y el tipo de afectación primaria (mús- culo, unión neuromuscular, nervios o motoneurona inferior). Los pacientes en la segunda década de la vida, edad en la que se desarrolla la adolescencia, presentan síntomas diferentes de la hipotonía o retraso en los hitos motores, propios de las ENM en edades mas tempranas. En este período es necesario mantener un alto nivel de sospecha clínica porque los signos y síntomas de las ENM pueden ser sutiles, de lenta evolución y no ser referidos directamente por el paciente afectado. Esta situación favorece el subdiagnóstico y diagnóstico tardío de estas afecciones. Conocer estos síntomas y signos favorece la sospecha y diagnóstico precoz así como un manejo y cuidados apropiados según cada patología. En esta revisión se hace én- fasis en el amplio espectro de la sintomatología que debe ser considerada cuando se sospecha la existencia de una ENM.
Neuromuscular disorders (NMD) include a multiplicity of different diseases with variable clinical symptoms according to age of presentation and type of primary involvement (muscle, neuromuscular junction, nerve or spinal motor neuron). Patients in their second decade of life, when adolescence arises, have distinctive symptoms different from hypotonia or motor milestone developmental delay, which are commonly seen in the early childhood. To be attentive to this clinical diversity is important in order to suspect a NMD, since occasionally the signs or symptoms can be subtle and potentially overlooked by the clinician until later in life. In this review we emphasize the broad spectrum of symptomatology that should be considered when suspecting a NMD during adolescence, to enhance the recognition of these pathologies and be aware of them for a better and earlier workup.
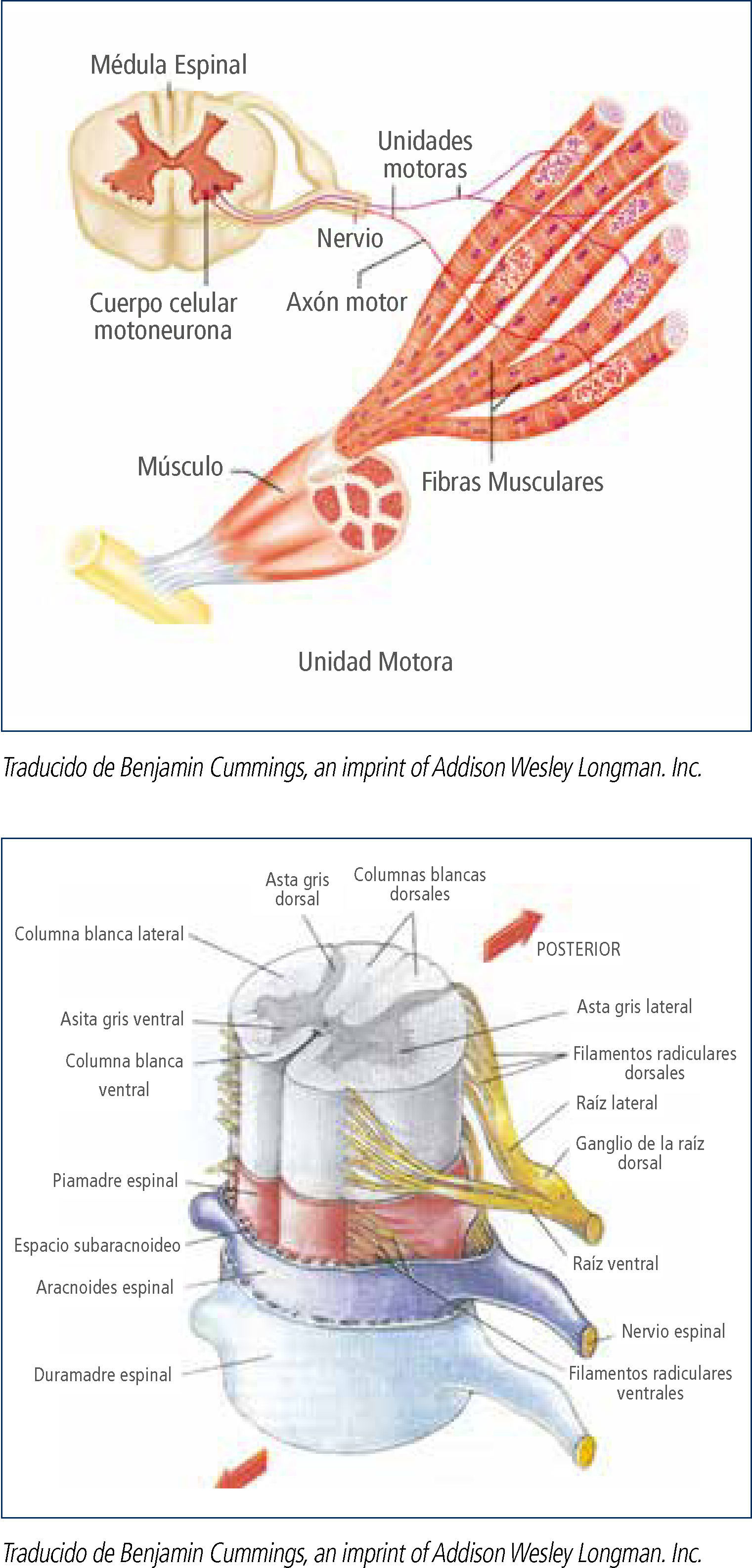
Las Enfermedades Neuromusculares (ENM) son un grupo de enfermedades caracterizadas por signos y síntomas secundarios al compromiso de algunos de los componentes de la unidad motora, es decir, la motoneurona inferior, el nervio periférico, la unión neuromuscular y el músculo (figura 1). La gran mayoría de ellas son enfermedades de origen genético, si bien las causas adquiridas: autoinmunes, inflamatorias o tóxicas, son también parte del espectro etiológico de estas afecciones. Los estudios de prevalencia de las ENM hereditarias indican una tasa global de 37/100.000 habitantes con una prevalencia de la Distrofia Miotónica (DM) de 10.6/100.000 habitantes, lo que representa un 28% del total, correspondiendo a la patología más frecuente en esta serie de enfermedades. Las distrofinopatías y la distrofia facio-escápulo-humeral (DFEH) la siguen en frecuencia con 22.9% y 10.7% respectivamente. La Atrofia Muscular Espinal (AME) representa un 5.1%, es decir 1.87/100.000 habitantes1. Una extrapolación de estas cifras a la población chilena proyectada por el Instituto Nacional de Estadística para el año 2013, (17.631.579 habitantes)2, permite calcular que en nuestro país existirían aproximadamente 1.860 pacientes portadores de DM; 1.490 con distrofinopatías; 690 pacientes con DFEH; 320 pacientes con AME. En relación a las neuropatías – los estudios de prevalencia de las neuropatías hereditarias han mostrado que la Enfermedad de Charcot-Marie-Tooth (CMT) es la enfermedad hereditaria más común del sistema nervioso periférico, con una prevalencia estimada de 1 en 1.214 personas3.

La estrategia diagnóstica para identificar estas enfermedades depende de la edad del paciente y de las manifestaciones clínicas iniciales, por esta razón, en este artículo se abordan estas patologías en la adolescencia, dado que en este período etario los signos y síntomas clínicos son menos evidentes que en edades mas precoces, requiriéndose de un alto índice de sospecha, una adecuada historia clínica y un examen físico dirigido, que busca elementos que permitan orientar un diagnóstico temprano y un tratamiento precoz. Es importante destacar que existe un grupo de pacientes que no consulta directamente por alguna dolencia atribuible fácilmente a una ENM, por lo que sólo un conocimiento adecuado del abanico de manifestaciones clínicas posibles permitirá un diagnóstico oportuno.
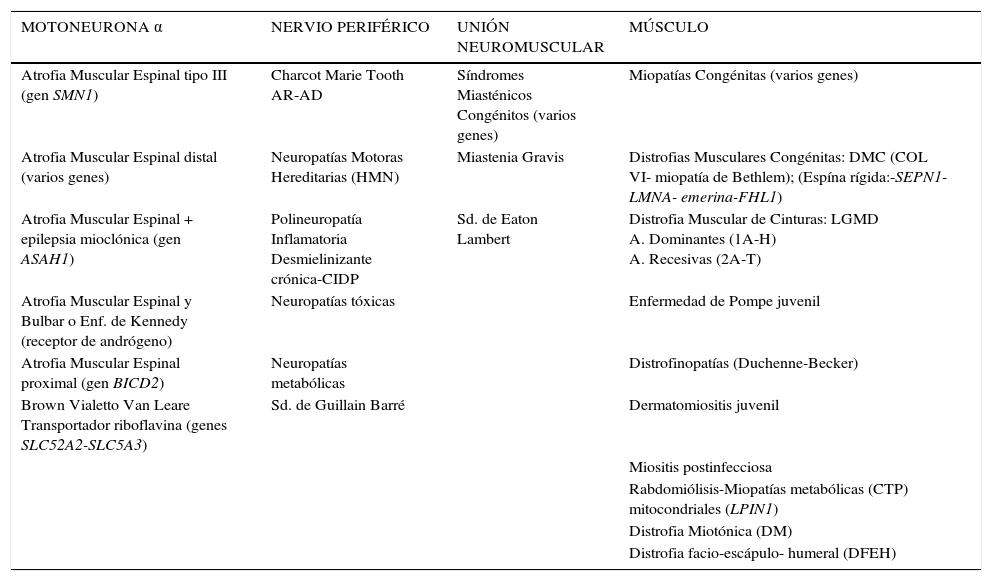
Las ENM que se presentan más comúnmente en la adolescencia se detallan en la tabla 1 En relación a los cuadros secundarios a compromiso de las motoneuronas, éstos varían desde una AME proximal tipo 3, por deleción del gen SMN1 hasta enfermedades mejor toleradas con una lenta evolución, como son algunas atrofias espinales distales que se presentan con debilidad y atrofia muscular distal de extremidades inferiores, en ocasiones desapercibida por años. Un tipo de AME asociada a epilepsia mioclónica progresiva se ha descrito recientemente, causada por una mutación recesiva en el gen ASAH1 que codifica para la enzima lisosomal ceramida ácida. Esta nueva entidad clínica contribuye a expandir mas aún el espectro de las enfermedades causadas por compromiso de la motoneurona4.
ENFERMEDADES NEUROMUSCULARES EN LA ADOLESCENCIA
| MOTONEURONA α | NERVIO PERIFÉRICO | UNIÓN NEUROMUSCULAR | MÚSCULO |
|---|---|---|---|
| Atrofia Muscular Espinal tipo III (gen SMN1) | Charcot Marie Tooth AR-AD | Síndromes Miasténicos Congénitos (varios genes) | Miopatías Congénitas (varios genes) |
| Atrofia Muscular Espinal distal (varios genes) | Neuropatías Motoras Hereditarias (HMN) | Miastenia Gravis | Distrofias Musculares Congénitas: DMC (COL VI- miopatía de Bethlem); (Espína rígida:-SEPN1-LMNA- emerina-FHL1) |
| Atrofia Muscular Espinal + epilepsia mioclónica (gen ASAH1) | Polineuropatía Inflamatoria Desmielinizante crónica-CIDP | Sd. de Eaton Lambert | Distrofia Muscular de Cinturas: LGMD A. Dominantes (1A-H) A. Recesivas (2A-T) |
| Atrofia Muscular Espinal y Bulbar o Enf. de Kennedy (receptor de andrógeno) | Neuropatías tóxicas | Enfermedad de Pompe juvenil | |
| Atrofia Muscular Espinal proximal (gen BICD2) | Neuropatías metabólicas | Distrofinopatías (Duchenne-Becker) | |
| Brown Vialetto Van Leare Transportador riboflavina (genes SLC52A2-SLC5A3) | Sd. de Guillain Barré | Dermatomiositis juvenil | |
| Miositis postinfecciosa | |||
| Rabdomiólisis-Miopatías metabólicas (CTP) mitocondriales (LPIN1) | |||
| Distrofia Miotónica (DM) | |||
| Distrofia facio-escápulo- humeral (DFEH) |
Algunas de las ENM posibles de observar en la segunda década de la vida.
Las enfermedades secundarias al compromiso de los nervios periféricos que se presentan en la adolescencia también presentan una gran diversidad.Destacan las neuropatías hereditarias sensitivo-motoras como el Charcot-Marie-Tooth (CMT) por su frecuencia3 y las neuropatías adquiridas como la Polineuropatía Inflamatoria Desmielinizante Crónica (CIDP), debido a la importancia de iniciar un tratamiento inmunomodulador que puede revertir el curso de la enfermedad5. Un pie cavo progresivo, la ausencia de reflejos osteotendíneos, asociado a una amiotrofia distal de las extremidades inferiores deben guiar la sospecha de un CMT; en cambio una debilidad subaguda proximal de extremidades con arreflexia y episodios recurrentes de debilidad simétrica debe hacer pensar en una CIDP. El Síndrome de Guillan-Barré, principal causa de parálisis fláccida aguda en individuos menores de 18 años, debe ser conocido con sus múltiples formas de presentación de manera de evitar investigaciones innecesarias e iniciar un tratamiento inmuno-modulador precoz. Las distrofias musculares de cinturas (LGMD), distrofinopatías, distrofia miotónica, DFEH, diversas miopatías congénitas, miopatías mitocondriales, metabólicas e inflamatorias, miotonías hereditarias, síndromes miasténicos, son todas patologías que pueden ser diagnosticadas también en la segunda década de la vida.
El objetivo de esta revisión es presentar los síntomas clínicos orientadores, los signos clínicos subyacentes y el compromiso de otros sistemas y órganos, distintos del músculoesquelético, que pueden acompañar a las diversas ENM que se presentan en este período de la vida.
SÍNTOMAS CLÍNICOS QUE ORIENTAN A LA EXISTENCIA DE UNA ENMTrastorno de la marcha. La mayor parte de las ENM indicadas anteriormente pueden presentar diversos grados de alteración de la marcha, como por ejemplo: bamboleo, fatigabilidad, asimetría, claudicación, marcha en punta de pies. La marcha bamboleante o anadina suele ser manifestación de una debilidad de la cintura pélvica por compromiso de los músculos proximales en las extremidades inferiores, especialmente de la musculatura glútea, tanto del glúteo mayor como del glúteo medio, ambos responsables de mantener la pelvis alineada. Durante la fase de apoyo unilateral de la marcha, este déficit hace evidente el bamboleo de la pelvis y su acentuación al requerir mayor velocidad al caminar. Esto es propio de la mayoría de las miopatías y distrofias musculares – aunque también puede verse en la Atrofias Musculares Espinales (AME) y en algunas miastenias congénitas. La existencia de fatigabilidad o cansancio fácil puede ser una manifestación secundaria a un síndrome miasténico en el que la transmisión neuromuscular a nivel de la placa motora está afectada y los síntomas de fatiga aparecen en la medida que se realiza ejercicio físico. Las miopatías metabólicas y las mitocondriales pueden manifestar fatiga por alteración de la producción energética del músculo. Se puede observar una marcha claudicante en algunas miopatías y neuropatías asimétricas, en la que el grado de compromiso de una extremidad es mayor que el contralateral, ocasionando cojera como en la DFEH y mononeuropatías. En el caso específico de la DFEH se debe considerar que muchas veces el motivo de consulta corresponde a la claudicación secundaria a la debilidad de la musculatura peronea y los signos de compromiso en cintura escapular pueden pasar desapercibidos en una primera instancia. La marcha en punta de pies no debe ser considerada como idiopática a esta edad. Este síntoma puede traducir un acortamiento del músculo tríceps-sural a nivel del Tendón de Aquiles, secundario a miopatías congénitas (central core, Bethlem, LGMD), miopatías distales6, neuropatías, (CMT) o distrofias musculares (Duchenne-Becker). Las miotonías congénitas tipo Becker o Thompsen pueden provocar alteración en la dinámica y fluidez de las fases de la marcha por las miotonías generalizadas desencadenadas por la actividad muscular al caminar.
Caídas frecuentes. Este síntoma se produce principalmente por debilidad muscular proximal o distal de las extremidades inferiores, donde el paciente pierde abruptamente la fuerza para mantener el equilibrio ya sea por fatiga o incapacidad de hacer uso de la musculatura apropiada frente a irregularidades del terreno o actividades que requieren de mayor fuerza como correr (AME-miopatías-distrofias musculares-sd. misténicos). El fenómeno de las caídas frecuentes se produce durante la fase de apoyo unilateral, en la cual todo el peso corporal debe ser asumido por una sola extremidad, lo cual genera fatiga o bien colapso de la cadena extensora (cuádriceps v/s glúteos) de dicha extremidad. El compromiso sensitivo, propioceptivo que acompaña a algunas neuropatías, puede afectar el input sensorial necesario para mantener el equilibrio y ocasionar también tendencia a caídas repetidas.
Dificultades para subir y/o bajar escaleras. Esta acción motora requiere de una considerable fuerza muscular, los glúteos asumen la propulsión del tronco en asociación directa con la musculatura extensora de tronco (paravertebrales) y el músculo cuádriceps, frecuentemente afectado en las ENM suele fatigarse y colapsar bruscamente. En los casos de mayor debilidad los pacientes refieren requerir tracción de las extremidades superiores para realizar esta maniobra. La existencia de una debilidad distal de extremidades inferiores principalmente de los músculos tibial anterior y peroneos, con la imposibilidad de realizar una flexión dorsal del pie (steppage) es otro mecanismo que dificulta subir y bajar el peldaño, ya que requiere de una gran compensación a nivel pélvico, reduciéndose las reacciones de equilibrio al estar en apoyo unilateral. Esta característica es propia de neuropatías, miopatías y AME de predominio distal.
Dolor muscular relacionado o no al ejercicio. El dolor durante o depués del ejercicio, en una intensidad mayor al dolor muscular propio de personas que no tienen entrenamiento físico, es un síntoma que debe llamar la atención del médico. El dolor de pantorrillas y muslos, en ocasiones también extremidades superiores, puede deberse a una miopatía metabólica como la Enfermedad de McArdle o glicogenosis tipo V, en la que se producen calambres musculares dolorosos, gatillados por contracciones musculares estáticas como cargar pesas o ejercicios dinámicos como correr. También algunas miopatías del metabolismo de los lípidos y la rabdomiólisis pueden ocasionar dolor7. Es importante recordar que tanto el Síndrome de Guillan-Barré como las miositis inflamatorias post infecciosas presentan dolor de las extremidades, en oportunidades bastante invalidante. Algunas neuropatías sensitivas de fibra pequeña pueden presentar dolor quemante e intenso en la planta de los pies como la Enfermedad de Fabry, por déficit de la enzima lisosomal alfa galactosidasa A8.
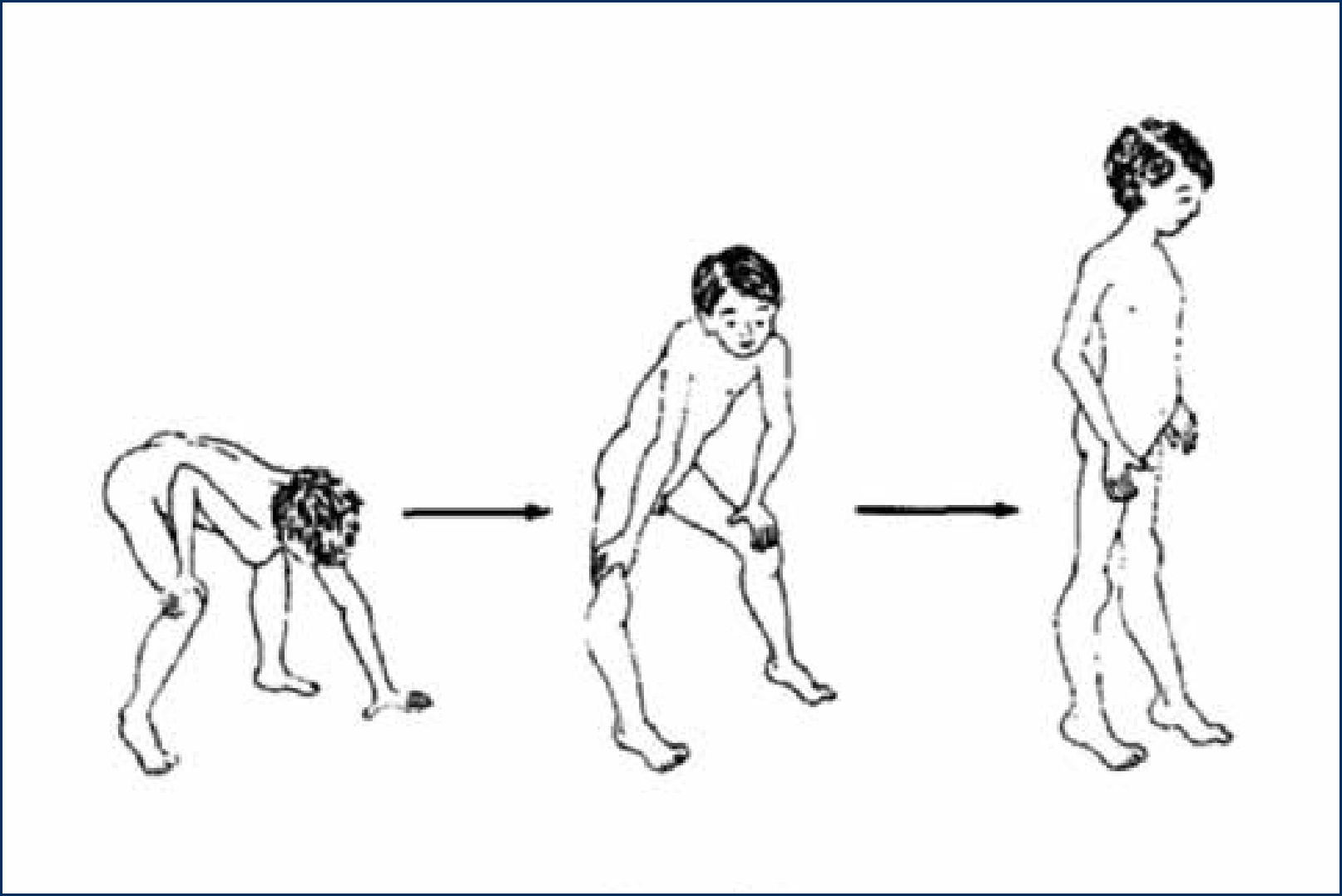
Dificultades para ponerse de pie desde el suelo. Este síntoma que acompaña a la debilidad muscular de la cintura pélvica se le conoce como Signo de Gowers. Los pacientes no logran levantarse desde el suelo sin apoyar una mano o ambas en el suelo y en sus propias piernas “trepando” por sobre sus propias extremidades inferiores con los brazos para lograr la posición de bipedestación (figura 2). Esta maniobra es una estrategia de los pacientes con debilidad de cuádriceps y glúteos, que les permite bloquear la rodilla, por tope óseo y conseguir la postura bípeda con un uso parcial de dicha musculatura.

Calambres. Los calambres suelen ser identificados como espasmos o contracturas musculares dolorosas y se observan en diversas miopatías y distrofias musculares, en particular en las miopatías metabólicas. Cuando las contracturas o espasmos no son dolorosos es importante identificar si nos encontramos frente a un fenómeno miotónico más propio de las distrofias miotónicas y de las miotonías congénitas. Entendemos por miotonía el retardo en la relajación de la musculatura esquelética posterior a una contracción voluntaria y puede buscarse dirigidamente en el examen físico del paciente.
Dificultades de alimentación, atoros. En los pacientes con Miastenia Gravis pueden presentarse con síntomas secundarios a un compromiso bulbar que ocasiona disfagia y atoros, pudiendo ser por algunos meses la única manifestación de una miastenia que se generalizará en el transcurso del tiempo. La Enfermedad de Pompe y la dermatomiositis pueden tener como manifestación inicial aislada la presencia de disfagia11,12 Existe una miopatía descrita recientemente caracterizada por debilidad diafragmática arreflexia, distress respiratorio y disfagia conocida con el epónimo de EMARDD13.
Infecciones respiratorias frecuentes y/o prolongadas. Este síntoma se debe a que un número importante de miopatías, distrofias musculares, miastenias congénitas y atrofias musculares espinales, presentan debilidad para toser y mantener despejada la vía aérea, asociado a la presencia de debilidad de los músculos respiratorios, diafragma e intercostales. Todos estos elementos contribuyen a la existencia de una capacidad ventilatoria disminuida e incluso una insuficiencia respiratoria restrictiva. La Enfermedad de Pompe, de comienzo mas tardío, es uno de los cuadros que se debe tener en consideración cuando nos enfrentamos a un paciente que tiene una discrepancia entre un severo compromiso respiratorio y la relativa mantención de la fuerzas de extremidades. La misma situación se observa en los síndromes miopáticos con espina rígida en los que el compromiso de la función respiratoria prevalece y es anterior a la debilidad de extremidades.
Trastornos del sueño. La disminución de la capacidad ventilatoria por las razones antes descritas, puede manifestarse inicialmente sólo por dificultades asociadas al sueño. Despertares nocturnos frecuentes, sensación de ahogo, ortopnea, cefalea matinal y cansancio durante el día. Todos estos son síntomas relacionados con una insuficiencia respiratoria inicial, hipoxia nocturna y retención de CO2. La distrofia miotónica constituye la ENM con la mayor tasa de trastornos del sueño, principalmente debido a la presencia de una somnolencia diurna excesiva pero también apneas centrales y obstructivas del sueño, síndrome de las piernas inquietas, entre otras. Estos trastornos del sueño están presentes aproximadamente en el 70-80% de los pacientes con DM14.
Visión doble. La parálisis o paresia de la musculatura extra ocular puede manifestarse inicialmente como diplopía. Esto es posible de observar en Miastenia Gravis, Síndrome de Miller Fisher, enfermedades mitocondriales entre otras.
Mioglobinuria. La mioglobinuria secundaria a una rabdomiólisis, ocasiona orinas oscuras por la presencia de mioglobina. Este es un síntoma de algunas miopatías metabólicas, también ocasionalmente de las distrofinopatías y más raramente en algunas distrofias musculares de cinturas como sarcoglicanopatías15. El déficit de carnitina palmitoil transferasa, (CPT) seguido por la deficiencia de maltasa ácida y la deficiencia de lipina, son la causa más común de rabdomiólisis aislada16.
SIGNOS DEL EXAMEN FÍSICO ASOCIADOS A DIVERSAS ENMDebilidad muscular. La falta de fuerza es el signo cardinal de las enfermedades que afectan la unidad motora. La evaluación segmentaria de las fuerzas se puede realizar fácilmente en adolescentes graduando el compromiso de fuerzas según grupos musculares y escala MRC (Medical Research Council) donde el esfuerzo del paciente se califica entre 0-5 (tabla 2), o bien con pruebas funcionales como saltar, correr, equilibrio unipodal, subir y bajar un escalón o realizar un cierto número de sentadillas.
DEBILIDAD MUSCULAR
| Grado 5: Músculo se contrae normalmente en contra de la resistencia máxima |
| Grado 4: La fuerza muscular está reducida, pero la contracción muscular aún se puede realizar contra la resistencia |
| Grado 3: La fuerza muscular se reduce aún más de tal manera que la articulación se puede mover sólo contra la gravedad sin la resistencia del examinador |
| Grado 2: Los músculos se pueden mover sólo si se elimina la resistencia de la gravedad |
| Grado 1: Sólo un atisbo de movimiento |
| Grado 0: No se observa ningún movimiento |
Escala para evaluación de fuerzas segmentarias de la Medical Research council.
Amiotrofia. El escaso desarrollo de la masa muscular o la destrucción de ésta, produce una amiotrofia generalizada en muchas miopatías, distrofias musculares y AME. Otros cuadros pueden presentar amiotrofias segmentarias como las miopatías distales, neuropatías y atrofias musculares distales en que la amiotrofia es de predominio distal de las extremidades inferiores.
Talla baja/bajo peso. Algunas ENM se caracterizan por presentar dificultades en el crecimiento, como las enfermedades mitocondriales y la condrodistrofia miotónica o Enfermedad de Schwartz-Jampel17.
Cambios de la piel. La dermatomiositis juvenil se acompaña de un eritema violáceo en los párpados, junto con lesiones típicas de calcinosis por depósitos de calcio, que aparecen como nódulos duros bajo la piel en nudillos de las manos (calcinosis). La Miopatía de Bethlem por mutación del gen que codifica para el colágeno VI de la matriz celular, produce con frecuencia queloides a veces incluso espontáneos o cicatrices como papel de cigarrillo. Este cuadro se presenta además con signos de hiperqueratosis folicular10.
Contracturas articulares progresivas. Las retracciones musculoesqueléticas se producen por fenómenos de destrucción y fibrosis muscular así como por la menor movilización de las articulaciones en pacientes con debilidad muscular. Las contracturas siempre se producirán en aquellas articulaciones en donde exista debilidad muscular y asimetría entre grupos musculares antogónicos. En la articulación de la rodilla se producen contracturas en flexión porque el cuádriceps es siempre más débil que los isquiotibiales. Se observan con frecuencia en algunos tipos de miopatías y distrofias musculares, fundamentalmente en la Miopatía de Bethlem y en la Distrofia Muscular de Emery Dreifuss, así como en distrofias y miopatías congénitas como las laminoapatías por mutaciones en el gen de la lamina A/C9.
En otras oportunidades entre los antecedentes del paciente identificamos la presencia al nacer de torticolis, pie bot, luxación de caderas que ayudarán a consolidar la hipótesis de una ENM. En particular las ENM causadas por mutaciones del colágeno VI (Enfermedad de Bethlem y Ulrich), las más frecuentes de las distrofias musculares congénitas10. Estas presentan la particularidad de que los pacientes pueden nacer con múltiples retracciones que van desapareciendo con el paso del tiempo.
Escoliosis progresiva. Es un fenómeno frecuente en las ENM, debido al compromiso de la musculatura paravertebral e intercostal que ocasiona debilidad axial con la consiguiente aparición de curvas escolióticas, que se rigidizan con el paso del tiempo. Las curvas siempre aparecen por una asimetría de la debilidad entre un hemicuerpo y otro o bien, por sobre uso de una de las extremidades.
Hipertrofia muscular. En las miotonías congénitas es posible observar un aumento generalizado de la musculatura esquelética, que otorga al paciente un aspecto atlético sin ser deportistas. La hipertrofia lingual se puede observar en las distrofinopatías, en las sarcoglicanopatías, distrofias musculares por mutación del gen relacionado con la fukutina (FKRP) y en la Enfermedad de Pompe. Una hipertrofia segmentaria de las pantorrillas es habitual en las distrofinopatías, en las sarcoglicanopatías, en las distrofias musculares relacionadas a mutaciones en el gen FKRP, y en ocasiones, en las formas mas tardías de atrofias musculares espinales (AME 3).
Rippling, fasciculaciones y miotonía. Se conoce como rippling la existencia de contracciones musculares eléctricamente silentes, que aparecen después de un estímulo mecánico o de estiramiento. Se observan sobre la superficie de los músculos ondulaciones que duran entre 5-20 segundos. Se observa en pacientes con mutaciones en el gen de la caveolina (CAV3). Las fasciculaciones son contracciones espontáneas de unidades motoras que se observan en pacientes con compromiso de las motoneuronas y otras condiciones que cursan con denervación. El fenómeno miotónico puede ser investigado solicitando al paciente que empuñe la mano con fuerza o cierre los párpados con fuerza. Se apreciará la dificultad en la relajación de la musculatura que ha sido contraída. Otra manera de observarla es percutiendo directamente los músculos donde se pude observar la aparición de una contracción sostenida.
Hiperlaxitud distal. Es un signo que acompaña clásicamente a la Miopatía de Bethlem en un inicio previo al desarrollo de contracturas, así como a la distrofia muscular e Ullrich, ambas patologías son secundarias a mutaciones de los genes que codifican para el colágeno VI.
Deformaciones torácicas. Pectum carinatum/excavatum. Este signo se observa en varias ENM y es secundario al compromiso y debilidad de la musculatura torácica18.
Pie cavo. Este es el clásico signo que se encuentra en la neuropatía de Charcot-Marie-Tooth, aunque se sabe actualmente que no todos los genes responsables de CMT se acompañan de pies cavos en su evolución. El pie cavo también puede presentarse en otras ENM, o puede ser constitucional. Por lo mismo siempre que se observe pie cavo debe investigarse su origen.
Ausencia reflejos osteotendíneos. La gran mayoría de las afecciones de la unidad motora se acompaña de reflejos osteotendíneos disminuidos o ausentes, con algunas excepciones representadas por algunos tipo de atrofias espinales distales o neuropatías motoras hereditarias en las que es posible encontrar reflejos rotulianos vivos19.
Luxaciones de la rótula. Este fenómeno se puede encontrar en pacientes con Miopatía de Bethlem por déficit del colágeno VI.
Temblor distal fino de manos-temblor de la lengua. El temblor fino distal de las manos secundario a la pérdida de las motoneuronas de las atrofias musculares espinales recibe el nombre de poliminimioclonus. El temblor de la lengua por la existencia de fasciculaciones puede observarse en estas mismas patologías.
Ptosis palpebral, oftalmoplegia. Las miastenias congénitas, la Miastenia Gravis, las miopatías mitocondriales y algunas miopatías congénitas suelen presentar ptosis palpebral y/o oftalmoplegia.
Paladar ojival. Este es un signo presente con frecuencia en las miopatías congénitas como miopatías nemalínicas, miotubulares, central core.
Voz nasal. Los trastornos de la voz se observan con frecuencia en diversas miopatías, atrofias musculares, neuropatías y miastenia. La voz puede ser hipofónica o de escaso volumen, por debilidad de la musculatura orofaríngea o disfónica o ronca en el caso de algunas neuropatías y atrofias espinales por paresia de las cuerdas vocales20.
COMPROMISO DE OTROS SISTEMAS QUE ACOMPAÑA A ALGUNAS ENMEl compromiso de otros órganos o tejidos es común en varias de las ENM. Los más importantes a tener en consideración son el compromiso cardíaco donde se pueden encontrar miocardiopatías dilatadas, restrictivas o alteraciones del ritmo cardíaco con arritmias y fibrilaciones ventriculares, entre otras. Pueden ser causa de muerte súbita en estos pacientes las laminopatías, por mutaciones en el gen de la lamina A/C, distrofinopatías, distrofias miotónicas, etc. En ocasiones el compromiso significativo del ritmo cardíaco requerirá de marcapaso o desfibriladores en forma preventiva para evitar una muerte súbita. La Enfermedad de Pompe en su forma infantil precoz se acompaña como signo cardinal de una insuficiencia cardiaca severa y letal.
El compromiso hepático con aumento de las transaminasas que se elevan y que pueden ser las musculares no por compromiso hepático como en DMD y disfunción hepática variable puede acompañar a algunas miopatías metabólicas como algunas glicogenosis o también a las enfermedades mitocondriales. Las miopatías inflamatorias por su compromiso multisistémico pueden presentar en su evolución un compromiso hepático.
En relación con aspectos endocrinológicos es importante destacar que la intolerancia a la glucosa, la resistencia a la insulina y la diabetes mellitus pueden ser complicaciones de la distrofia miotónica y de algunas miopatías, como las laminopatías y las miopatías por mutación en el gen de la selenoproteína (SEPN1)21. Un hipotiroidismo y/o hipoparatiroidismo puede observarse en algunas de las miopatías mitocondriales. La ginecomastia, que puede ser asimétrica, es un signo inicial frecuente en la Enfermedad de Kennedy (atrofia muscular bulbo espinal ligada al X) en la adolescencia junto con dolores musculares y fatiga22.
Compromiso del SNc. La Distrofia Muscular de Duchenne presenta un compromiso cognitivo variable, desde grados menores hasta retardo mental y conductas autistas. La Distrofia Miotónica se acompaña con frecuencia de alteraciones cognitivas variables. Gran parte de las Distrofias Musculares Congénitas (DMC) por defectos de la glicosilación de las proteínas y cuyo marcador es un déficit de alfa-distroglicano en la membrana de la fibra muscular, pueden presentar malformaciones de la corteza cerebral y cerebelo, trastornos de la migración neuronal, ventriculomegalias, quistes cerebelosos, entre otros. La DMC por déficit de merosina presenta en las imágenes de Resonancia Magnética una leucoencefalopatía con alteraciones de la substancia blanca e hiperintensidad en secuencias ponderadas en T2 de las regiones subcorticales y periventriculares.
Una epilepsia mioclónica progresiva puede observarse en una forma deAME, recientemente descrita4 y en varias de las enfermedades mitocondriales.
El compromiso ocular como cataratas, retinopatías – microftalmia acompaña a algunos pacientes con DM-Miopatías mitocondriales, DMC.
El compromiso auditivo con hipoacusia o sordera observar en las DFEH de inicio muy precoz y en miopatías mitocondriales. Algunas neuropatías también pueden presentar sordera.
El compromiso gastrointestinal es una constipación crónica frecuente de observar en varias de las enfermedades musculares con fecalomas, dilatación gástrica aguda y cólicos, entre otros.
Datos significativos de la historia personal y familiar del paciente
Frente a la presencia de algunos de los síntomas o signos descritos anteriormente, es importante completar la historia clínica del paciente con la información de los antecedentes personales y familiares. Una consanguinidad se encuentra con frecuencia entre los antecedentes de pacientes con ENM hereditarias recesivas y no debemos olvidar indagar sobre este aspecto. ¿Hay antecedentes de familiares afectados por cuadros semejantes?¿Hay antecedente de muerte súbita en la familia? ¿Cardiopatías o arritmias? ¿Presentó el paciente retraso en la adquisición de hitos motores o una marcha tardía? ¿Tuvo al nacer pie bot? ¿Luxación de caderas? ¿Artrogriposis? ¿Tortícolis? ¿Hay antecedente de escoliosis en la familia? ¿Queloides? ¿Hipertermia maligna con la anestesia? Realizar una adecuada historia clínica es fundamental para llegar a un diagnóstico adecuado.
CONCLUSIONESEl objetivo de esta revisión de los síntomas y signos más frecuentemente observados en los adolescentes con ENM, es contribuir a mejorar la sospecha diagnóstica precoz que permita indicar un tratamiento y manejo oportuno de los mismos.
Como una manera de ejemplificar la complejidad progresiva de este campo de la medicina, basta con señalar que actualmente las miopatías congénitas conforman un grupo de más de 30 trastornos musculares hereditarios, causados por 20 genes distintos, con signos de presentación clínica semejantes, cuyo diagnóstico específico continúa siendo un desafío. Asimismo las neuropatías hereditarias son otro ejemplo de la complejidad genética y fenotípica que han alcanzado muchas de las ENM, con 47 genes conocidos actualmente, sólo se logra identificar alguna mutación en el 50% de los pacientes con CMT3. La tabla genética de Kaplan23 que se encarga de mantener un registro actualizado de todas las ENM monogénicas refiere la existencia de 685 fenotipos diferentes; 365 genes; 28 de los cuales codifican para alguna de las proteínas mitocondriales.
La sospecha diagnóstica de una ENM debe realizarse precozmente y orientar al paciente hacia unidades de diagnóstico y tratamiento que cuenten con equipos multidisciplinarios que permitan la realización de la complejidad de exámenes de laboratorio, neuroimágenes, biopsia muscular, estudios electrofisiológicos y genéticos que estas patologías requieren. La oportuna indicación de un tratamiento que considere todas la prevención de las eventuales complicaciones junto a un consejo genético a la familia, forman parte del manejo necesario en todo paciente con una enfermedad neuromuscular.
Los autores declaran no tener conflictos de interés, en relación a este artículo.