



Las distrofias musculares congénitas (DMC) son enfermedades musculares hereditarias muy heterogéneas. Su diagnóstico se basa en criterios histológicos (signos distróficos en músculo) y clínicos (de inicio neonatal o durante la infancia precoz, antes de adquirir la marcha). Los lactantes presentan hipotonía, movilidad disminuida y retraso o estancamiento del desarrollo motor. Son enfermedades progresivas, con frecuente aparición de retracciones articulares, rigidez y deformidad de columna, insuficiencia respiratoria y, en algunas formas, afectación cardiaca. Por ello, necesitan un manejo multidisciplinario neuro-ortopédico, cardio-respiratorio y frecuentemente quirúrgico. Los valores de las enzimas musculares (CK) permiten orientar el diagnóstico. Formas con aumento marcado de CK guían hacia la DMC con déficit de merosina (gen LAMA2) y las distroglicanopatías, muy heterogéneas genéticamente. Ambas pueden asociar anomalías cerebrales y retraso mental (frecuente en las distroglicanopatías), o limitarse a la sustancia blanca cerebral, sin deterioro cognitivo (merosinopatías). La biopsia muscular ayuda en el diagnóstico detectando el déficit de merosina o la glicosilación anómala de distroglicano. Las formas de DMC sin aumento importante de CK no presentan afectación cerebral y se diagnostican frecuentemente gracias a la identificación de signos clínicos como hiperlaxitud distal en la DMC de Ullrich (genes COL6), rigidez espinal selectiva y fallo respiratorio en las selenopatías (gen SEPN1) o “drop-head” en las laminopatías (gen LMNA). La resonancia magnética muscular es muy útil para confirmar la sospecha clínica y orientar casos difíciles. La identificación genética y el desarrollo de modelos animales están ayudando a conocer mejor la fisiopatología de estas enfermedades, aumentando las posibilidades de encontrar terapias para el futuro.
Congenital muscular dystrophies (CMDs) are inherited muscle disorders with a high clinical and genetic heterogeneity. Diagnostic criteria are histological (dystrophy in muscle biopsy) and clinical (onset at birth or before the age of walking). Affected infants typically appear “floppy” with low muscle tone and poor spontaneous movements. Patients may present with delay or arrest of gross motor development. CMDs show progressive weakness, joint contractures, spinal stiffness and deformities, respiratory compromise and certain types may develop cardiac symptoms. Multidisciplinary management is required, including neuro-orthopedic, cardio-respiratory and often surgical treatment. Dosage of muscle enzymes (CK) helps in the diagnostic pathway. Marked increase is observed in merosin deficient CMD (LAMA2 gene) and in the dystroglycanopathies, which are genetically very heterogeneous. Both disorders may show central nervous system abnormalities, being often associated with mental retardation in dystroglycanopathies, while merosinopathies show only striking white matter changes and intellect is not impaired. Muscle biopsy helps to detect defects in merosin expression or abnormal dystroglycane glycosylation. CMDs without marked increase of CK do not associate brain involvement. Diagnosis is often oriented by the identification of clinical markers such as distal hyperlaxity in Ullrich CMD (COL6 genes), selective spinal rigidity and respiratory failure in selenopathies (SEPN1 gene), or dropped head in laminopathies (LMNA gene). Muscle MRI is very useful to confirm clinical suspicion and orientate difficult cases. Gene identification in these disorders and development of animal models are helping to better understand their physiopathologic mechanisms, opening the possibility for future therapies.
Las distrofias musculares congénitas (DMCs) son una familia de enfermedades hereditarias raras muy heterogéneas clínica y genéticamente. Los estudios epidemiológicos estiman una prevalencia entre 0.68 y 2.5 por 100000 habitantes1. La frecuencia relativa de los diferentes tipos de DMC varía geográficamente, concentrándose algunas formas en ciertas regiones del mundo debido a la existencia de mutaciones ancestrales, como por ejemplo la DMC de tipo Fukuyama en Japón2 o muscle-eye–brain en Finlandia3. Junto con las miopatías congénitas, las DMCs son las miopatías estructurales más frecuentes en la infancia. Los criterios diagnósticos de DMC fueron descritos en el siglo pasado, antes de la disponibilidad de diagnóstico genético y son fundamentalmente clínicos (inicio temprano, antes de la edad de la marcha) e histológicos (aspecto distrófico del músculo, sin características morfológicas específicas de las miopatías congénitas) (Figura 1)4,5. Los avances moleculares y el mejor conocimiento de estas enfermedades en las últimas décadas han mostrado que existe una superposición clínica e histológica con otras miopatías precoces, en particular con ciertas miopatías congénitas6, y un espectro continuo con las distrofias de cinturas (LMGD 2I)7 o con enfermedades musculares inicialmente descritas de forma separada en el adulto, como la distrofia de Emery-Dreifuss8 o la miopatía de Bethlem9.
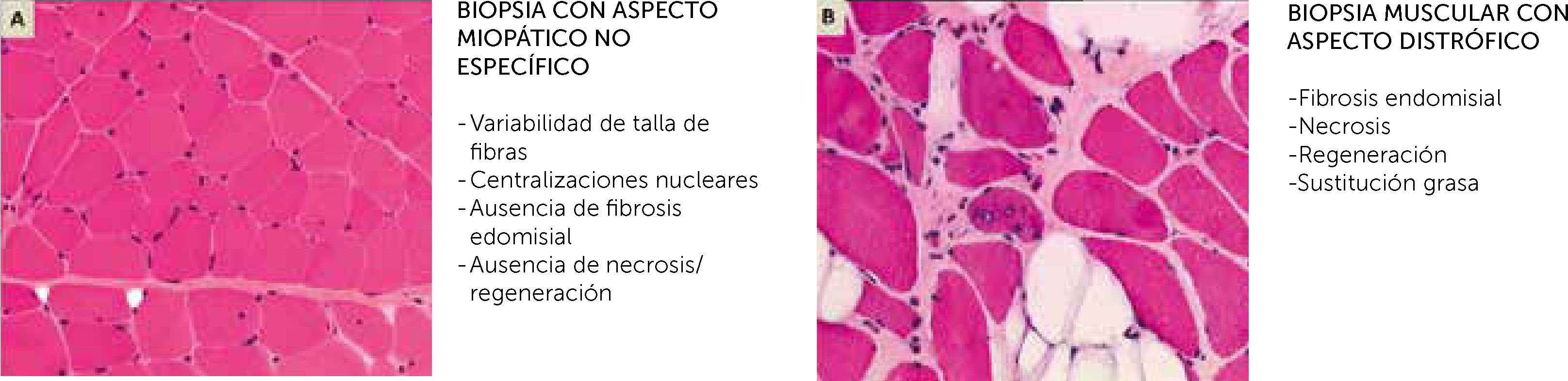
 Aspecto de una miopatía no distrófica, que puede mostrar muestra alteraciones (variabilidad de talla de fibras, centralizaciones nucleares) pero no contiene anomalías distróficas (fibrosis endomisial, fibras en necrosis o regeneración, sustitución de músculo por adipocitos (B). En realidad, las dos imágenes corresponden a dos biopsias musculares de una paciente de 14 años con mutaciones en el gen SEPN1. Las dos biopsias fueron realizadas con 3 meses de diferencia, una a nivel del músculo deltoides (A) y el otra en un músculo abdominal, en el curso de una cirugía de columna (B). La biopsia del músculo deltoides muestra anomalías no específicas de una miopatía congénita (A), mientras que la biopsia de músculo axial es muy distrofia (B) (Cortesía de A. Ferreiro).")
Biopsia muscular, estudio de microscopio óptico con tinción Hematoxilina-Eosina
(A) Aspecto de una miopatía no distrófica, que puede mostrar muestra alteraciones (variabilidad de talla de fibras, centralizaciones nucleares) pero no contiene anomalías distróficas (fibrosis endomisial, fibras en necrosis o regeneración, sustitución de músculo por adipocitos
(B). En realidad, las dos imágenes corresponden a dos biopsias musculares de una paciente de 14 años con mutaciones en el gen SEPN1.
Las dos biopsias fueron realizadas con 3 meses de diferencia, una a nivel del músculo deltoides (A) y el otra en un músculo abdominal, en el curso de una cirugía de columna (B). La biopsia del músculo deltoides muestra anomalías no específicas de una miopatía congénita (A), mientras que la biopsia de músculo axial es muy distrofia (B) (Cortesía de A. Ferreiro).
Las características fenotípicas, la fisiopatología y los defectos genéticos de las DMCs son muy variables, con más de 25 genes diferentes conocidos hasta el momento.
El diagnóstico diferencial de las DMCs se realiza principalmente con otras patologías neuromusculares precoces (miopatías congénitas, síndromes miasténicos congénitos, miopatías metabólicas, atrofia espinal), pero también existen enfermedades no neuromusculares a tener en cuenta en el lactante hipotónico o con retraso motor, como el sindrome de Prader –Willy10.
CLASIFICACIÓNLa denominación de las diferentes formas de miopatías y distrofias musculares hereditarias es compleja, debido a su gran heterogeneidad y al hecho de que muchas fueron inicialmente descritas de forma separada en base a diferentes características clínicas o inmunohisto-químicas. La identificación posterior de las causas genéticas y el mejor conocimiento de sus mecanismos fisiopatológicos ha conducido a la reclasificación de algunas de ellas o la pérdida de fronteras nosológicas. Por ejemplo, las selenoproteinopatías fueron inicialmente identificadas como una forma de DMC con sindrome de columna rígida11,12, para luego descubrir que también conducían a la forma clásica de miopatía multi-minicore6. La genética ha permitido también comprender el espectro de algunos fenotipos, aproximando entidades descritas anteriormente de forma separada. Es el caso del sindrome de Ullrich y la miopatía de Bethlem9, que no son sino extremos de un espectro continuo de una patología con presentación y severidad variable (colagenopatías VI).
Recientemente, para simplificar la clasificación y facilitar la comprensión de las miopatías estructurales hereditarias, se ha propuesto la denominación según el nombre del gen implicado, seguido de “-RD” (related dystrophy) o “-RM” (relates myopathy), según el gen esté clásicamente relacionado a una distrofia muscular o no (p.e SEPN1-related myopathy)1.
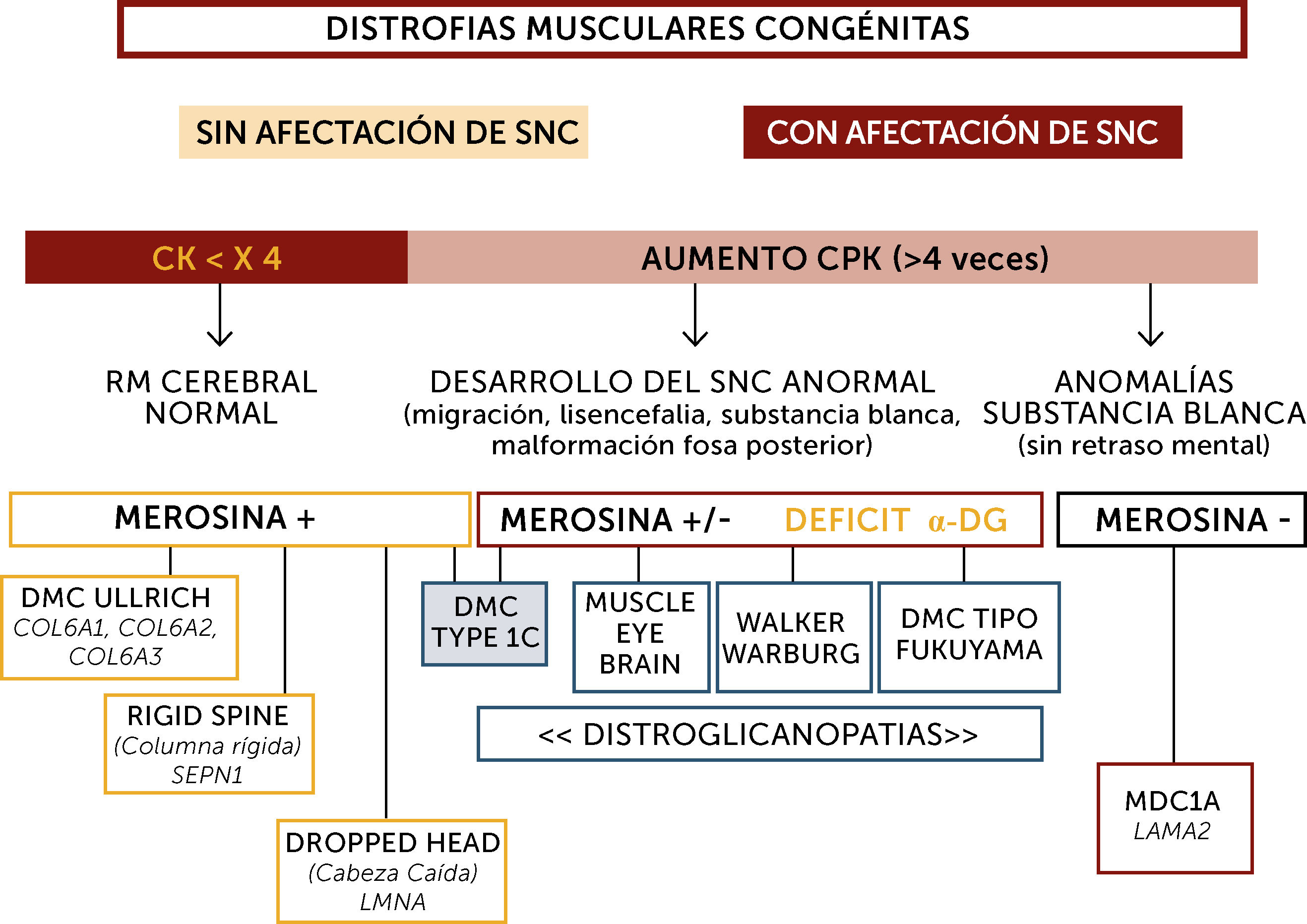
En la práctica, la clasificación más sencilla para el diagnóstico de las DMCs es la basada en las características fenotípicas definidas por criterios clínicos, exámenes complementarios, y análisis inmuno-histoquímicos y genéticos10 (Figura 2). Así, la presencia o no de retraso mental (Figuras 3 y 4), del aumento o no de los niveles de enzimas musculares (CK), de la aparición o no de anomalías neuro-radiológicas y de la expresión de diferentes proteínas (merosina, distroglicano, colágeno), permiten habitualmente orientar el diagnóstico.
 principales La orientación diagnóstica de las DMC se hace en base a la afectación o no del sistema nervioso central (SNC), el nivel de enzimas musculares (normal o no muy elevado; marcadamente elevado), datos neuroradiológicos (RM cerebral normal, con anomalías estructurales o de la substancia blanca aisladas), inmunohistoquímicos (expresión de merosina y alfa distroglicano) y marcadores fenotípicos (hiperlaxitud distal, rigidez espinal, pérdida de sostén cefálico, hipertrofia muscular, sindrome cerebro-ocular (“Muscle-eye-brain” (MEB)). Sindrome de Walker-Warburg, DMC de tipo Fukuyama). Los pacientes inteligentes, con RM cerebral normal suelen tener nivel de CK normal o poco aumentadas, expresión de merosina normal y pueden tener tres tipos de DMC por mutación de los genes SEPN1, COL6 o LMNA fundamentalmente. Las formas con retraso mental o con aumento marcado de CKs pueden tener formas con déficit primario o secundario en merosina. Los déficits primarios son debidos a mutaciones del gen LAMA2 y se caracterizan por un respeto de las funciones cognitivas pero anomalías difusas de la substancia blanca cerebral. Los déficits de merosina secundarios tienen un déficit de glicosilación de alfa distroglicano (distroglicanopatías). El espectro de fenotipos de las distroglicanopatías es muy extenso, desde pacientes sin retraso mental, hasta otros con retraso y malformaciones cerebro-oculares o de fosa posterior, o anomalías de substancia blanca supratentorial de extensión variable).")
Algoritmo diagnóstico global de las distrofias musculares congénitas (DMC) principales
La orientación diagnóstica de las DMC se hace en base a la afectación o no del sistema nervioso central (SNC), el nivel de enzimas musculares (normal o no muy elevado; marcadamente elevado), datos neuroradiológicos (RM cerebral normal, con anomalías estructurales o de la substancia blanca aisladas), inmunohistoquímicos (expresión de merosina y alfa distroglicano) y marcadores fenotípicos (hiperlaxitud distal, rigidez espinal, pérdida de sostén cefálico, hipertrofia muscular, sindrome cerebro-ocular (“Muscle-eye-brain” (MEB)). Sindrome de Walker-Warburg, DMC de tipo Fukuyama). Los pacientes inteligentes, con RM cerebral normal suelen tener nivel de CK normal o poco aumentadas, expresión de merosina normal y pueden tener tres tipos de DMC por mutación de los genes SEPN1, COL6 o LMNA fundamentalmente. Las formas con retraso mental o con aumento marcado de CKs pueden tener formas con déficit primario o secundario en merosina. Los déficits primarios son debidos a mutaciones del gen LAMA2 y se caracterizan por un respeto de las funciones cognitivas pero anomalías difusas de la substancia blanca cerebral. Los déficits de merosina secundarios tienen un déficit de glicosilación de alfa distroglicano (distroglicanopatías). El espectro de fenotipos de las distroglicanopatías es muy extenso, desde pacientes sin retraso mental, hasta otros con retraso y malformaciones cerebro-oculares o de fosa posterior, o anomalías de substancia blanca supratentorial de extensión variable).
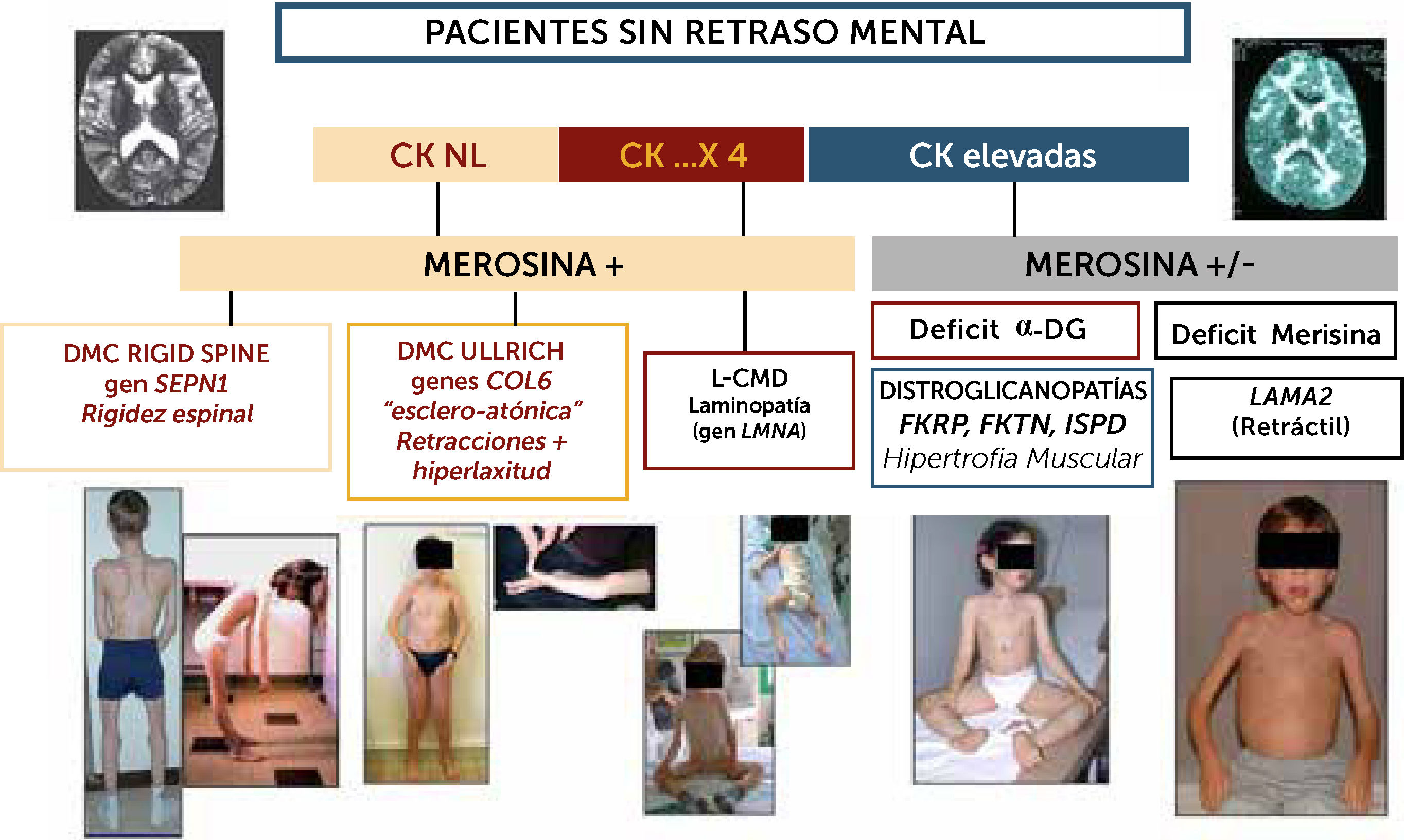
, expresión inmuno-histoquímica (merosina, alfa-DG). Los marcadores clínicos son particularmente útiles para sospechar las diferentes formas: en las formas con CK poco elevadas o normales, si el paciente presenta una rigidez selectiva de la columna deberá estudiarse el gen SEPN1 en prioridad (rigid spine sindrome); si existe una hiperlaxitud distal junto con una rigidez axial y retracciones articulares proximales (hombros, codos, caderas rodillas) se sospechara primeramente una DMC de tipo Ullrich y los genes a estudiar serán COL6A1, COL6A2 y COL6A3). Si el paciente no tiene progresos motores o pierde el sostén cefálico se sospechará una Laminopatía (gen LMNA); En casos de CKs elevadas hay dos posibilidades fundamentalmente: si el fenotipo es retráctil, con músculos más bien atróficos, inteligente pero con anomalías difusas en RM de substancia blanca cerebral, el diagnóstico más probable será de DMC con déficit de merosina. Si el fenotipo es hipertrófico, una distroglicanopatía por uno de los tres genes del subgrupo de déficit de fosfato-inositol deberán ser estudiados (FKRP, FKTN, ISPD).")
Algoritmo diagnóstico de las formas de DMC sin retraso cognitivo significativo
La orientación diagnóstica de las DMC en el niño sin retraso cognitivo puede ser guiada por datos biológicos (CK), expresión inmuno-histoquímica (merosina, alfa-DG). Los marcadores clínicos son particularmente útiles para sospechar las diferentes formas: en las formas con CK poco elevadas o normales, si el paciente presenta una rigidez selectiva de la columna deberá estudiarse el gen SEPN1 en prioridad (rigid spine sindrome); si existe una hiperlaxitud distal junto con una rigidez axial y retracciones articulares proximales (hombros, codos, caderas rodillas) se sospechara primeramente una DMC de tipo Ullrich y los genes a estudiar serán COL6A1, COL6A2 y COL6A3). Si el paciente no tiene progresos motores o pierde el sostén cefálico se sospechará una Laminopatía (gen LMNA); En casos de CKs elevadas hay dos posibilidades fundamentalmente: si el fenotipo es retráctil, con músculos más bien atróficos, inteligente pero con anomalías difusas en RM de substancia blanca cerebral, el diagnóstico más probable será de DMC con déficit de merosina. Si el fenotipo es hipertrófico, una distroglicanopatía por uno de los tres genes del subgrupo de déficit de fosfato-inositol deberán ser estudiados (FKRP, FKTN, ISPD).
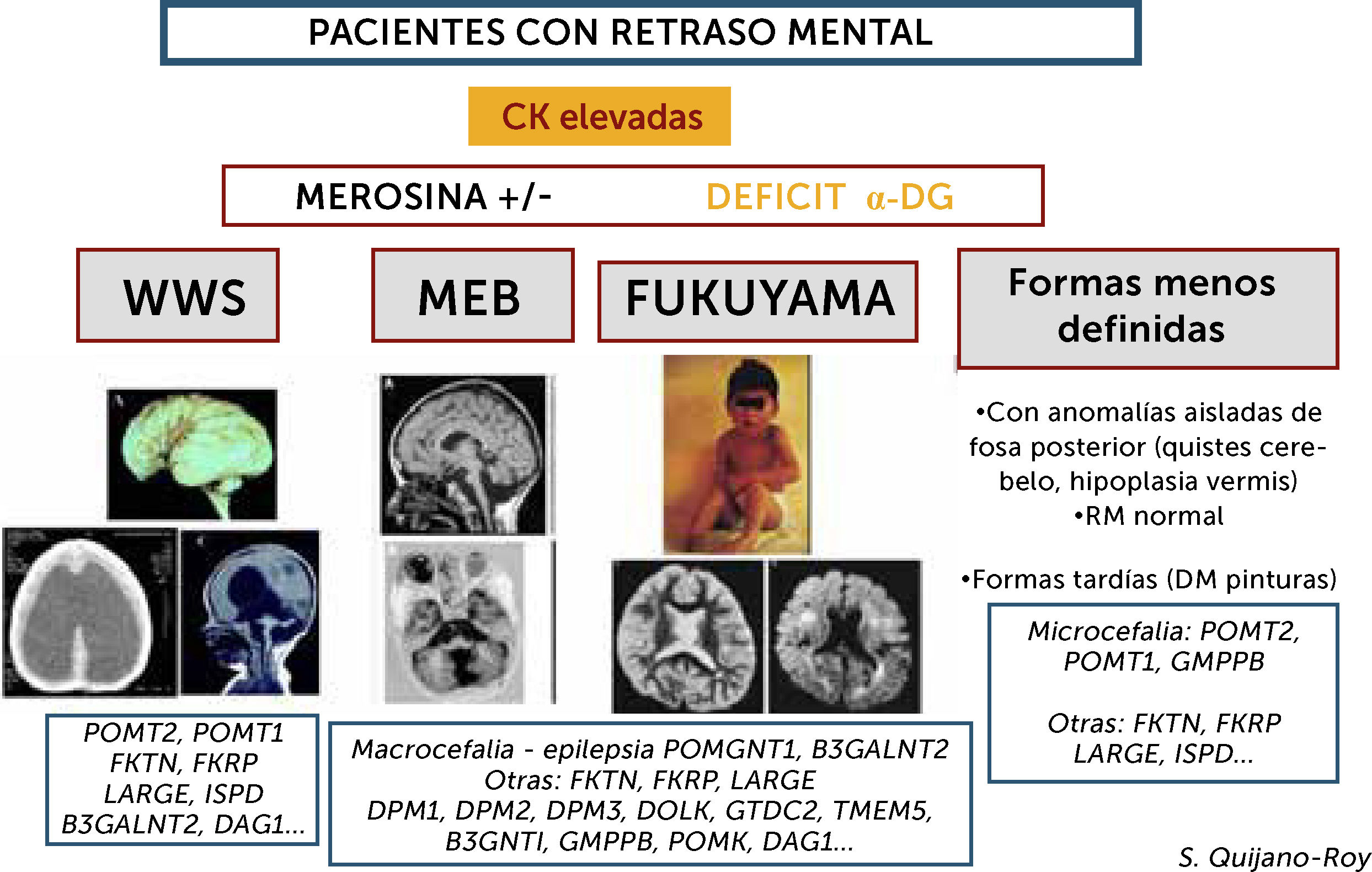
 Este grupo de DMC presenta incremento importante de CKs de forma constante (salvo si estado muy avanzado de la enfermedad con atrofia muscular severa) y pueden presentar varios tipos de síndromes: Walker-Warburg (WWS), con lisencefalia de tipo empedrado (cobblestone) y anomalías muy severas malformativas de cerebro fosa posterior y tronco cerebral, incompatibles con la vida o con pronóstico de supervivencia limitado. Muscle-Eye-Brain (MEB) que presenta anomalías menos severas múltiples (malformaciones en cerebro, cerebelo y tronco, anomalías de substancia blanca) con frecuencia asociadas a afectación ocular. Esta forma en Japón se debe fundamentalmente a un gen (FKTN o fukutina, y conduce a la DMC de tipo Fukuyama). Se pueden encontrar, además, casos con RM normal y con anomalías mínimas (microcefalia) o aisladas de fosa posterior, o vermis cerebeloso, que no son conocidas por términos específicos. Hay actualmente al menos 18 genes, existiendo una heterogeneidad genética de los diferentes síndromes.")
Algoritmo de las DMCs con retraso mental (distroglicanopatías)
Este grupo de DMC presenta incremento importante de CKs de forma constante (salvo si estado muy avanzado de la enfermedad con atrofia muscular severa) y pueden presentar varios tipos de síndromes: Walker-Warburg (WWS), con lisencefalia de tipo empedrado (cobblestone) y anomalías muy severas malformativas de cerebro fosa posterior y tronco cerebral, incompatibles con la vida o con pronóstico de supervivencia limitado. Muscle-Eye-Brain (MEB) que presenta anomalías menos severas múltiples (malformaciones en cerebro, cerebelo y tronco, anomalías de substancia blanca) con frecuencia asociadas a afectación ocular. Esta forma en Japón se debe fundamentalmente a un gen (FKTN o fukutina, y conduce a la DMC de tipo Fukuyama). Se pueden encontrar, además, casos con RM normal y con anomalías mínimas (microcefalia) o aisladas de fosa posterior, o vermis cerebeloso, que no son conocidas por términos específicos. Hay actualmente al menos 18 genes, existiendo una heterogeneidad genética de los diferentes síndromes.
Se pueden distinguir cinco formas principales de DMCs:
1• Merosinopatías o DMC merosin negativa (MDC1A), debida a mutaciones en el gen LAMA2, que codifica la cadena alfa 2 de la proteína laminina 2 (o merosina).
2• Colagenopatías VI o DMC de tipo Ullrich (UCMD), causada por mutaciones en alguno de los 3 genes del colágeno VI (COL6A1, COL6A2 y COL6A3).
3• Distroglicanopatías, producidas por un trastorno de la glicosilación del alfa-distroglicano, con formas sindrómicas que incluyen retraso mental o anomalías estructurales cerebrales (tipo Fukuyama, tipo Muscle-Eye-Brain, tipo Walker-Warburg) o sin retraso mental (MDC1C), causadas por al menos 18 genes diferentes.
4• Selenoproteinopatías o sindrome de la columna rígida de tipo 1 (RSMD1), debido a la mutación del gen de la selenoproteína N1 (SEPN1).
5• Laminopatías congénitas (L-CMD), vinculadas al gen de la lámina A/C (LMNA).
Aunque las frecuencias de estas formas varían geográficamente, las más frecuentes en la mayoría de los países son las merosinopatías, las colagenopatías y las distroglicanopatías. Las DMCs presentan una herencia autosómica recesiva, excepto las laminopatías (L-CMD) y un porcentaje importante de sindromes de Ullrich (UCMD), en los cuales se pueden observar mutaciones dominantes de novo (un porcentaje menor se heredan de forma recesiva). Las formas de DMC también pueden clasificarse en función de la localización subtisular o subcelular de la proteína mutada y/o sus consecuencias a nivel de la fibra del músculo esquelético (matriz extracelular, sarcolema, lámina basal, retículo endoplasmático y envoltura nuclear).
FORMAS PRINCIPALES DE DMCDMC por déficit primario en merosina (MDC1A)Es una forma muy frecuente, afectando a entre 20-40% de los casos de DMC identificados en la mayoría de los países13,14. Se debe a mutaciones en el gen LAMA2 (en 6q22) que codifica la cadena alfa 2 laminina, una de las cadenas que forman laminina-2 o merosina15. La merosina es una proteína heterodimérica de la matriz extracelular que se encuentra en el músculo esquelético, la piel, las células de Schwann y la placenta. El modo de transmisión es autosómico recesivo. El diagnóstico se confirma mediante el estudio del gen LAMA2, un gen de gran tamaño (64 exones, 9.4 Kb). Dada la existencia en la mayoría de casos de mutaciones privadas, la interpretación de los resultados puede ser compleja. Las nuevas técnicas moleculares (secuenciación a alto débito) facilitan la identificación de mutaciones.
El diagnóstico de MDC1A se sospecha ante la presentación clínica, la elevación constante y franca de la CK (>4 veces lo normal) y la presencia de cambios distróficos en músculo asociados a un déficit de la expresión sarcolémica de los anticuerpos contra la cadena alfa 2 de la merosina16. Existe un espectro continuo entre la forma clásica de déficit total de merosina y las formas más atenuadas de déficit parcial, cuyos primeros signos pueden observarse durante la segunda década17. Dado el gran tamaño de la proteína deficitaria, los estudios inmunohistoquímicos pueden requerir la utilización de al menos dos anticuerpos diferentes (80kDa y 300kDa), sobre todo en casos de déficit parcial, pues en estos pacientes puede no detectarse el déficit con un solo anticuerpo (Figura 5). El estudio de la merosina también se puede realizar a partir de una muestra de piel y en los estudios prenatales a partir de vellosidades coriónicas. Es importante señalar que los pacientes con distroglicanopatías pueden presentar una expresión disminuida de la merosina (déficit secundario) ya que los residuos glicosilados de la proteína distroglicano sirven para anclar la cadena alfa 2 de la merosina y ésta disminuirá su unión en caso de déficit de glicosilación18.
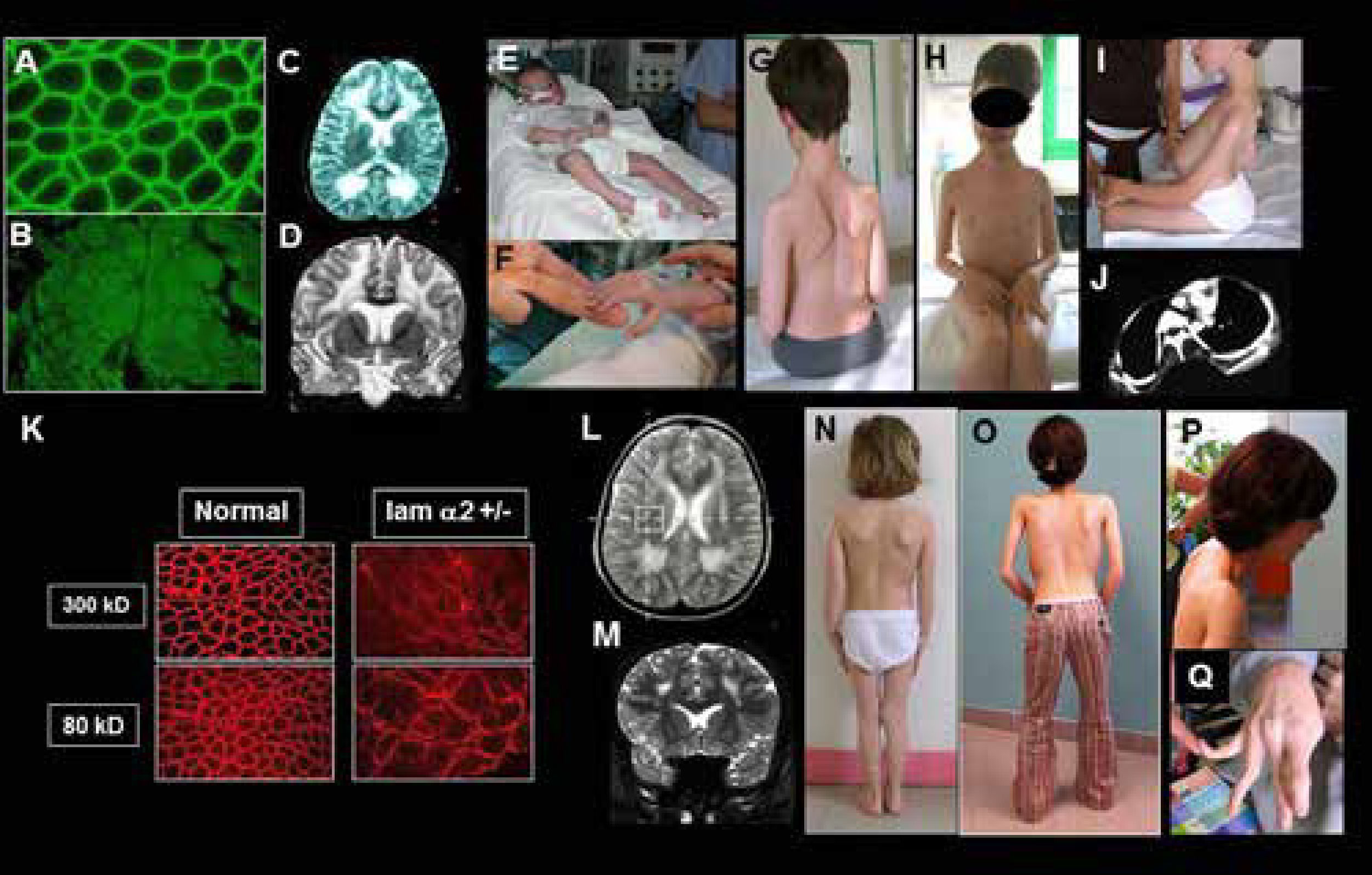
 o déficit parcial (K-P) A-B) Biopsia muscular con normalidad de expresión de la cadena alfa 2 de la laminina 2 (merosina) (A); expresión ausente (déficit complete) (B); C-D) Resonancia magnética cerebral con secuencias en T2, en cortes axiales (C) y frontales (D) que muestran una anomalía de substancia blanca difusa, con normalidad del cuerpo calloso y capsula interna. E-F) Paciente con déficit completo (clásico): inicio precoz severo, requiriendo ventilación asistida al nacimiento. El paciente desarrollo retracciones articulares muy precoces en miembros superiores e inferiores. G-I) Pacientes sin adquisición de la marcha y signos típicos del déficit completo en merosina: aspecto macrocefálico, retracciones articulares difusas y rigidez espinal. Paciente tratado desde edad temprana con corsé (G-H) paciente con lordo- escoliosis (I), paciente con complicaciones respiratorias y ortopédicas progresivas debido a la lordosis torácica con compresión bronquial del cuerpo vertebral. K-Q) Paciente con deficiencia parcial en merosina (ambulante): K) Resultados inmuno-histoquimicos usando dos anticuerpos, que muestran ausencia de expresión (300 Kda) y expresión parcial (80 Kda). L-M) RM cerebral en T2, cortes axiales (L) y frontales (M) que muestran anomalias de la substancia blanca más leves que en los pacientes con déficit completo y sin adquisición de la marcha. N-Q) Características clínicas de tres pacientes con déficit discreto en merosina. La rigidez espinal y las contracturas de codos son particularmente marcadas (O, P) y existe una hiperlaxitud de los dedos de las manos, que puede confundir con el fenotipo de colagenopatia o DMC de Ullrich (Q). (Quijano-Roy et al. Congenital Muscular Distrophies, Neuromuscular Imaging - Wattjes & Fischer, Springer, 1st Ed, 2013: 180).")
DMC de tipo merosina deficiente por mutaciones del gen LAMA2. Resultados inmunohistoquímicos y características clínicas en casos con déficit completo (A-J) o déficit parcial (K-P)
A-B) Biopsia muscular con normalidad de expresión de la cadena alfa 2 de la laminina 2 (merosina) (A); expresión ausente (déficit complete) (B);
C-D) Resonancia magnética cerebral con secuencias en T2, en cortes axiales (C) y frontales (D) que muestran una anomalía de substancia blanca difusa, con normalidad del cuerpo calloso y capsula interna.
E-F) Paciente con déficit completo (clásico): inicio precoz severo, requiriendo ventilación asistida al nacimiento. El paciente desarrollo retracciones articulares muy precoces en miembros superiores e inferiores. G-I) Pacientes sin adquisición de la marcha y signos típicos del déficit completo en merosina: aspecto macrocefálico, retracciones articulares difusas y rigidez espinal. Paciente tratado desde edad temprana con corsé (G-H) paciente con lordo- escoliosis (I), paciente con complicaciones respiratorias y ortopédicas progresivas debido a la lordosis torácica con compresión bronquial del cuerpo vertebral.
K-Q) Paciente con deficiencia parcial en merosina (ambulante): K) Resultados inmuno-histoquimicos usando dos anticuerpos, que muestran ausencia de expresión (300 Kda) y expresión parcial (80 Kda). L-M) RM cerebral en T2, cortes axiales (L) y frontales (M) que muestran anomalias de la substancia blanca más leves que en los pacientes con déficit completo y sin adquisición de la marcha. N-Q) Características clínicas de tres pacientes con déficit discreto en merosina. La rigidez espinal y las contracturas de codos son particularmente marcadas (O, P) y existe una hiperlaxitud de los dedos de las manos, que puede confundir con el fenotipo de colagenopatia o DMC de Ullrich (Q). (Quijano-Roy et al. Congenital Muscular Distrophies, Neuromuscular Imaging - Wattjes & Fischer, Springer, 1st Ed, 2013: 180).
Aunque la mayoría de los pacientes descritos con mutaciones LAMA2 no presentan deterioro intelectual, existe una anomalía muy llamativa de la sustancia blanca supratentorial, visible en las secuencias T2, Flair19 o STIR, sobre todo a partir de los 6 meses (antes, la anomalía puede no ser evidente debido a la inmadurez fisiológica de la mielinización). Estudios en resonancia magnética espectroscópica y difusión (DTI) indican un edema vasogénico de la substancia blanca. La merosina se localiza en la lámina basal vascular y su déficit conduciría a un aumento de la permeabilidad. La señal es normal en áreas en las que se compacta más mielina (cuerpo calloso, cerebelo, cápsula interna)20. Un pequeño porcentaje de pacientes presentan clínica cerebral. La epilepsia, en forma de convulsiones parciales complejas (con o sin generalización secundaria) de fácil control con antiepilépticos habituales es relativamente frecuente y puede dar síntomas sutiles (ausencias, pérdidas de conciencia o mareos inexplicados). Los casos de epilepsia refractaria y o con retraso mental son mucho más raros y pueden mostrar en RM anomalías estructurales supra e infratentoriales, especialmente polimicrogiria o agiria cerebral occipital1. Por otro lado, las técnicas de neurofisiología (potenciales evocados, EMG) muestran signos de daño a la mielina central y periférica que permanecen pauci o clínicamente asintomáticos21. Además de las complicaciones descritas, hay descripciones de afectación cardiaca de ritmo o de función ventricular izquierda sobre todo en la edad adulta, recomendándose un seguimiento con ecografía y Holter ECG de 24 horas de forma regular.
La presentación y severidad clínica varía según la deficiencia de merosina sea total o parcial22 (Figura 5). Los niños con un déficit total de merosina muestran un inicio precoz y severo, con hipotonía y debilidad global, trastornos de succión – deglución y fatiga o complicaciones respiratorias que pueden requerir soporte ventilatorio. No hay adquisición de la marcha. Se trata de una miopatía muy retráctil, que conduce a deformidades y retracciones progresivas en prácticamente todas las articulaciones de los miembros y cinturas (escapular, pélvica), de la columna e incluso de articulación temporo-mandibular. Por ello, se debe instaurar precozmente una fisioterapia de estiramiento intensivo articular y cervico-axial (al menos 3 sesiones semanales) y el tratamiento ortopédico. Son útiles las ortesis y corsés, que no impidan la función respiratoria (p. e. corsé de Garches)1,23. La columna suele evolucionar a una lordo-escoliosis torácica, con riesgo de compresión bronquial por lo que es importante realizar controles anuales radiológicos de la columna (con radiografías de frente y perfil) o completar con scanner torácicos en caso de agravación rápida de la función respiratoria. El tratamiento quirúrgico es con frecuencia necesario para estabilizar la escoliosis, ya sea mediante artrodesis al final del crecimiento o con las nuevas técnicas sin fusión, que permiten operar en periodos de crecimiento24,25. Las dificultades respiratorias son inicialmente debidas a la fatiga y debilidad respiratoria, bulbar (dificultades de deglución, atragantamientos, aspiraciones) y del tubo digestivo (reflujo gastroesofágico). Con los años, la retractilidad y rigidez de la caja torácica conduce a una insuficiencia respiratoria restrictiva severa, necesitando soporte ventilatorio de forma constante antes de la edad adulta. Debe explorarse la respiración durante el sueño anualmente o bianualmente cuando la capacidad vital esté disminuida, sobre todo por debajo de 60% de los valores teóricos, con el fin de detectar la necesidad de instaurar una ventilación. En nuestra experiencia, la utilización diaria de tratamientos fisioterapéuticos con presión positiva desde la pequeña infancia (máquinas de tipo IPPB o intermittent positive presure Breathing) puede retrasar la necesidad de ventilación, que suele ser necesaria antes de la edad adulta. En caso de necesitar durante el día mediante traqueotomía o mediante ventilación intermitente a demanda por medio de una pipeta. La atrofia muscular es constante en estas formas precoces y severas, pero puede existir en la evolución una hipertrofia de la lengua. Los trastornos alimentarios son frecuentes debido a la debilidad oro-facial y bulbar, los trastornos de la motilidad digestiva y las complicaciones respiratorias y de ortodoncia, contribuyendo a los trastornos del crecimiento. La afectación de los músculos óculomotores puede observarse desde la primera década, sobre todo un déficit de la mirada vertical y lateral. Es frecuente la aparición trastornos digestivos crónicos (estreñimiento, reflujo). En la evolución, muchos pacientes presentan un aspecto macrocefálico.
Cuando el déficit de merosina no es completo (déficit parcial), la severidad de las complicaciones motrices, ortopédicas y respiratorias suele ser menor, con posibilidad de adquisición de la marcha y sin necesidad de ventilación durante la edad pediátrica en los que no presentan pérdida de la marcha. Puede existir una hipertrofia de gemelos. La aparición de escoliosis debe ser vigilada porque puede evolucionar muy rápidamente y agravar la función respiratoria. A veces, la presentación puede ser engañosa, con una rigidez de la columna vertebral o un déficit de cinturas, más sugestivo de distrofia de cinturas o de distrofia de Emery-Dreifuss.
DMC de tipo Ullrich (UCMD)El sindrome clínico descrito por Ullrich en 1930 o distrofia esclero-atónica26 representa la forma más grave de colagenopatía del músculo esquelético, constituyendo alrededor del 20% de entre todas las DMC en los países occidentales14. En Japón es la segunda forma de DMC, tras la de tipo Fukuyama.
La DMC de tipo Ullrich (UCMD) es secundaria al déficit de colágeno VI, que es un heterotrímero de proteínas esencial para las funciones de la matriz extracelular en el músculo esquelético y la piel. El colágeno VI está compuesto de tres cadenas diferentes bajo el control de tres genes distintos, COL6A1, COL6A2 y COL6A327. La miopatía de Bethlem, diagnosticada fundamentalmente en adultos, es una forma alélica de UCMD más retráctil y menos severa a nivel motor y respiratorio, vinculada también al colágeno VI por la mutación de uno de estos mismos tres genes. Existe un continuo clínico entre estas dos miopatías9. Las mutaciones encontradas en diferentes genes COL6 pueden ser recesivas o dominantes (60% dominantes de novo) y existen algunas mutaciones recurrentes relativamente frecuentes. En casos con anomalías no descritas, la confirmación del diagnóstico genético y el asesoramiento pueden ser complejos, siendo indispensable para ello disponer de las muestras de los progenitores.
La presentación clínica clásica es la de un lactante o niño con debilidad y fatigabilidad muscular relativamente difusa, asociadas a una hiperlaxitud articular distal marcada, que contrasta con la presencia de retracciones de las articulaciones proximales y axiales, a menudo tempranas y especialmente progresivas28. La hipotonía, la debilidad muscular y las retracciones pueden estar presentes desde el nacimiento (tortícolis, luxación de la cadera, cifosis congénita), o aparecer posteriormente, causando un retraso en el desarrollo motor, caídas frecuentes y posturas anormales de los miembros debido a la hiperlaxitud o a las deformaciones articulares y espinales. No hay afectación cardíaca ni del sistema nervioso central (inteligencia y resonancia magnética cerebral normales). Dependiendo del nivel motor máximo alcanzado, se pueden distinguir varios grupos de pacientes: aquellos que nunca adquieren la marcha (severos precoces), los que adquieren la marcha pero la pierden antes de la edad adulta (moderados progresivos), y los que mantienen la marcha hasta la edad adulta (leves) (Figura 6). Estos últimos podrían ser también clasificados como miopatía de Bethlem precoz, sobre todo cuando presentan retracciones distales28. En contraste con la DMC merosin negativa, el grupo más numeroso de pacientes con UCMD adquiere la deambulación, aunque la mayoría pierden esta capacidad antes de la edad adulta. La hiperlaxitud articular va disminuyendo con el tiempo a nivel axial y proximal, y en casos avanzados queda limitada a regiones distales de los miembros (falanges de manos y de pies). A nivel de los miembros superiores es típica la postura con caída de manos y dedos largos y finos de consistencia disminuida. La protrusión del calcáneo, la piel rugosa por hiperqueratosis y las cicatrices queloides son muy frecuentes y permiten en muchos casos orientar el diagnóstico. En contraste con otros síndromes con hiperlaxitud como los síndromes Ehler-Danlos (forma cifoescoliótico por mutaciones del gen FKBP14), los pacientes con DMC de Ullrich presentan muy precozmente una rigidez espinal y limitaciones de la movilidad articular que se agravan progresivamente, afectando primero a las cinturas (escapular, caderas) para luego extenderse a las grandes articulaciones de los miembros (codos, rodillas) y finalmente a las regiones distales (muñecas, tobillos, dedos). En casos leves o incipientes, las retracciones deben buscarse sobre todo a nivel de la abducción de las caderas y hombros, de la extensión de los codos y de la extensión de las falanges de los dedos de la mano. Esta exploración puede realizarse pidiendo al paciente que una las manos juntas en posición de plegaria, poniendo en evidencia la imposibilidad de extender los dedos (Figura 6). La rigidez de la columna vertebral con cifosis o cifoscoliosis necesita un seguimiento y tratamiento fisioterapéutico y ortopédico adaptado intensivo, similar a la DMC merosin deficiente. Igualmente, la DMC de Ullrich se asocia en la evolución a una insuficiencia respiratoria restrictiva con una caja torácica rígida29. Los pacientes con sindrome de Ullrich presentan además típicamente una disfunción del diafragma30, que puede ponerse en evidencia comparando las pruebas de función respiratoria en posiciones diferentes (disminución en decúbito supino). Por esta razón, la vigilancia y manejo respiratorio y ortopédico deben ser incluso más precoces y frecuentes que en la forma merosina negativa. En general, los pacientes que no adquieren la marcha o que la pierden durante la infancia necesitan ventilación nocturna antes de la edad adulta.
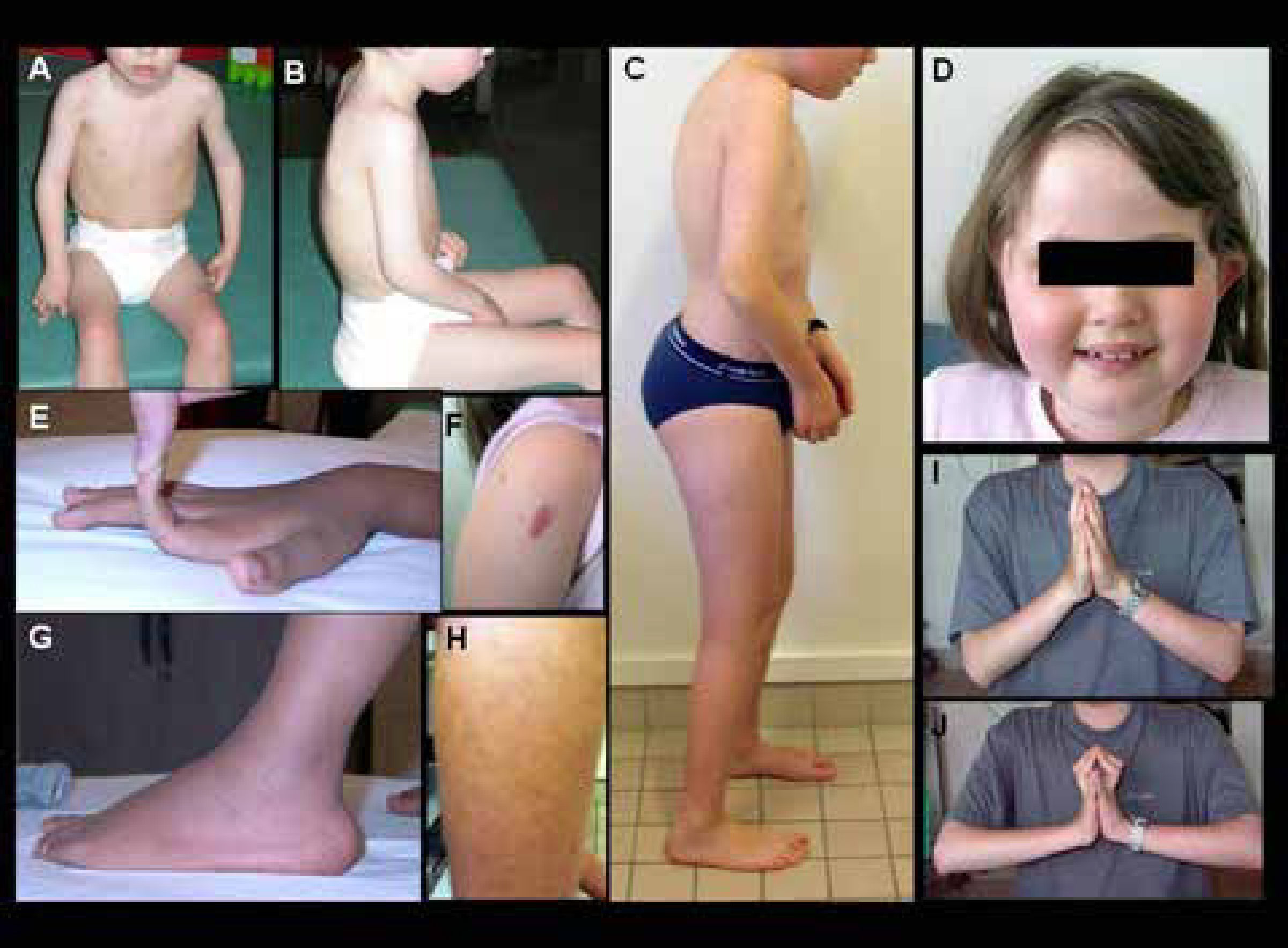
 Niño con forma precoz severa (sin adquisición de la marcha). Hay una cifosis dorsal rigida y retracciones en codos y miembros inferiores. C) Forma moderada-severa (con adquisición de la marcha pero con agravación progresiva en la infancia), que muestra signos similares pero menos marcados. D-J) Diferentes marcadores fenotípicos caracteristicos de esta forma, incluyendo facies redondeada y cuello fino (D), hiperlaxitud distal de dedos (E), hipertrofia de cicatrices (F), protrusión de calcáneos (G), piel rugosa o granulosa (H), y retracciones de las articulaciones de los dedos detectadas cuando se pide al paciente que coloque sus manos en postura de “plegaria” con extensión máxima de las muñecas (J comparado a I). (Quijano-Roy et al. Congenital Muscular Distrophies, Neuromuscular Imaging - Wattjes & Fischer, Springer, 1st Ed, 2013: 189)")
Características clínicas de pacientes con DMC de Ullrich
A-B) Niño con forma precoz severa (sin adquisición de la marcha). Hay una cifosis dorsal rigida y retracciones en codos y miembros inferiores. C) Forma moderada-severa (con adquisición de la marcha pero con agravación progresiva en la infancia), que muestra signos similares pero menos marcados. D-J) Diferentes marcadores fenotípicos caracteristicos de esta forma, incluyendo facies redondeada y cuello fino (D), hiperlaxitud distal de dedos (E), hipertrofia de cicatrices (F), protrusión de calcáneos (G), piel rugosa o granulosa (H), y retracciones de las articulaciones de los dedos detectadas cuando se pide al paciente que coloque sus manos en postura de “plegaria” con extensión máxima de las muñecas (J comparado a I). (Quijano-Roy et al. Congenital Muscular Distrophies, Neuromuscular Imaging - Wattjes & Fischer, Springer, 1st Ed, 2013: 189)
El diagnóstico a menudo se evoca fácilmente en presencia de los marcadores clínicos descritos anteriormente y de las características evolutivas. A diferencia del déficit en merosina, las CK son normales o ligeramente elevadas (menos de 4 veces los valores normales). La resonancia magnética muscular se ha convertido en una herramienta de diagnóstico muy interesante, por la detección de un patrón de afectación específico, tanto por los músculos afectados como por el tipo de lesiones observadas. Éstas tienen un aspecto en bandas, visibles en algunos músculos, como por ejemplo en el muslo (recto femoral, vasto lateral), pierna (banda entre sóleo y gastrocnemios), brazo (tríceps), hombro (deltoides) (Figura 7)31-34. Estas bandas pueden no ser evidentes en pacientes muy jóvenes o con fenotipos poco severos. El análisis de la biopsia muscular puede mostrar desde signos miopáticos no específicos hasta un patrón distrófico obvio. Una marcada ausencia o disminución de colágeno VI en la inmunohistoquímica de la biopsia muscular es un marcador valioso para el diagnóstico, pero inconstante, sobre todo en caso de mutación dominante, ya que la expresión de colágeno VI puede estar relativamente preservada gracias a la expresión del alelo normal. La existencia de tres genes implicados y la posibilidad de mutaciones dominantes y recesivas para cada uno de ellos, hace que este paso sea extremadamente difícil y complejo28. Hasta épocas recientes, el diagnóstico de esta forma solía ser orientado mediante estudios de secreción de colágeno VI en cultivo de fibroblastos de la piel, que pueden detectar la retención citoplasmática de colágeno no secretado y orientar el análisis genético de una de las cadenas si existe una disminución selectiva de transcritos28,9. Actualmente, con las nuevas técnicas moleculares, la búsqueda de anomalías es más directa y rápida. Aun así, en casos de mutaciones no repertoriadas y de carácter patogénico dudoso, la interpretación puede ser difícil y necesitar estudios complementarios. En los últimos años se han identificado mutaciones intrónicas en algunos casos típicos que no habían mostrado mutaciones en los análisis clásicos de secuenciación de exones.
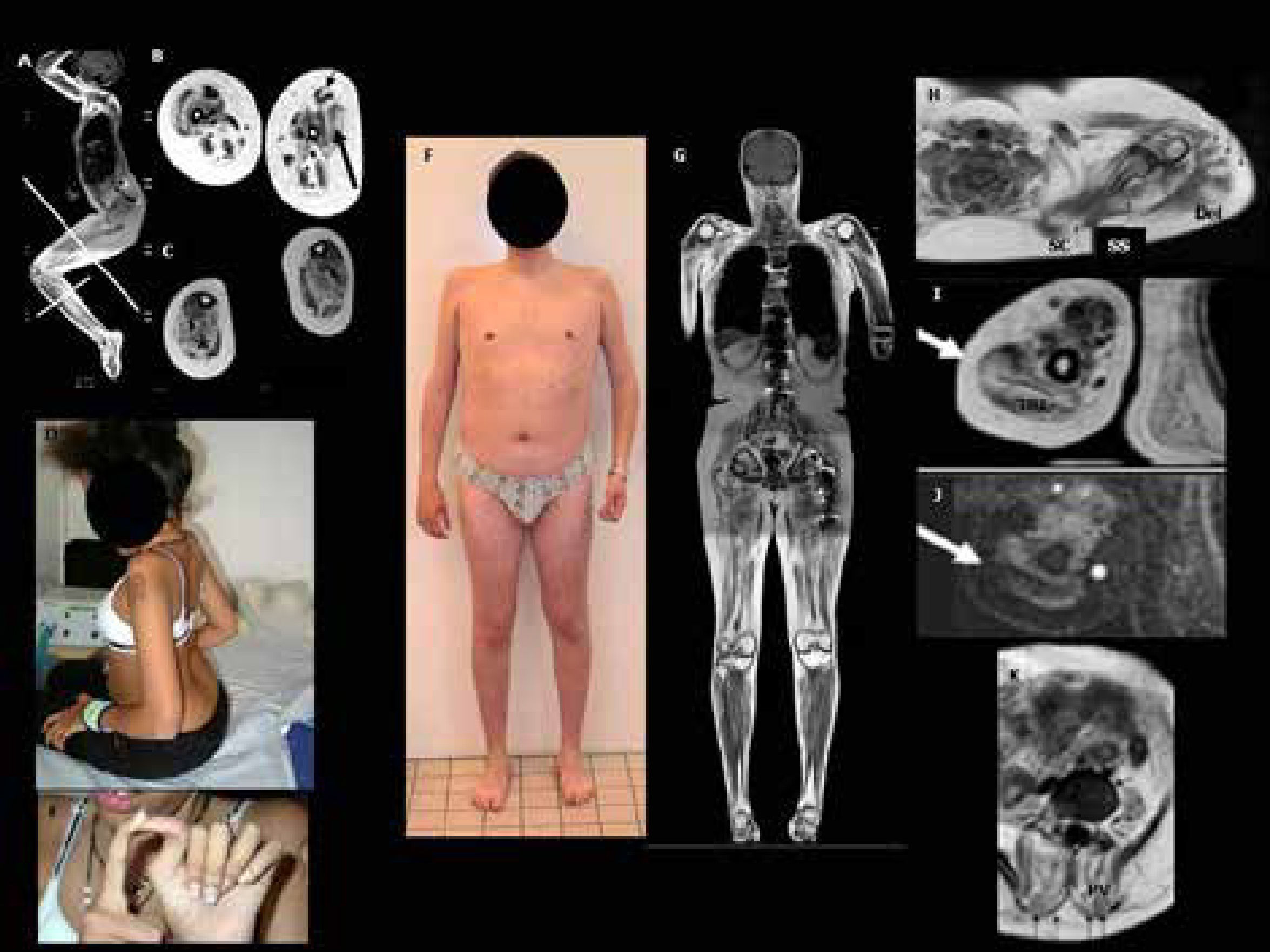
 Forma intermediaria Ullrich/Bethlem (F-K) A-C: RM muscular de cuerpo entero (T1-TSE) en una niña de 12 años con UCMD, que perdió la marcha a los 10 años de edad. A) Debido a las retracciones articulares severas de caderas y rodillas, un protocolo y posicionamiento especial deben realizarse en el examen de RM con el fin de obtener imágenes con cortes de muslos y piernas perpendiculares al eje mayor de las extremidades. A notar el patrón típico de RM muscular de la miopatía COL6 en miembros inferiores. B) RM a nivel del muslo que muestra las bandas típicas en bizcocho enrollado a nivel del músculo vasto lateral (flecha gruesa negra) y en el recto anterior que presenta una muesca vertical (flechas finas). C) La pierna muestra el borde ribeteado híper-intenso entre el músculo soleo y los gastrocnemios (estrellas negras). D,E) Aspecto clínico del paciente con retracciones de codos y cicatriz hipertrofiada (D), e hiperlaxitud llamativa de los dedos(E). F-K: Características clínicas y patrón radiológico de un paciente de 21 años con una forma intermediaria (Ullrich/Bethlem). F) Inicio de síntomas en la infancia con retraso de marcha e hiperlaxitud, evolución lentamente progresiva con desarrollo de retracciones de codos y tobillos durante la infancia. H-K) La RM muscular de cuerpo entero muestra el aspecto típico en bandas (tigroide), en el que se alternan bandas de híper e hipo intensidad (flechas) en diferentes músculos de diferentes regiones: H) Hombros: bandas radiales en el músculo deltoides (Del), perpendiculares en el músculo subescapular (SC) y supra espinoso (SS). I-J) Imagen de RM del brazo, en secuencia T1-TSE (I) y STIR (J): banda horizontal en el músculo tríceps braquial (TRI). K) Signos de infiltración fibro-adiposa severa y bandas verticales en los músculos paravertebrales lumbares (PV). (Quijano-Roy et al. Congenital Muscular Distrophies, Neuromuscular Imaging - Wattjes & Fischer, Springer, 1st Ed, 2013: 190).")
Aspecto clínico y RM muscular en pacientes con miopatías relacionadas con COL6: DMC Ullrich moderada progresiva (A-E) Forma intermediaria Ullrich/Bethlem (F-K)
A-C: RM muscular de cuerpo entero (T1-TSE) en una niña de 12 años con UCMD, que perdió la marcha a los 10 años de edad. A) Debido a las retracciones articulares severas de caderas y rodillas, un protocolo y posicionamiento especial deben realizarse en el examen de RM con el fin de obtener imágenes con cortes de muslos y piernas perpendiculares al eje mayor de las extremidades. A notar el patrón típico de RM muscular de la miopatía COL6 en miembros inferiores. B) RM a nivel del muslo que muestra las bandas típicas en bizcocho enrollado a nivel del músculo vasto lateral (flecha gruesa negra) y en el recto anterior que presenta una muesca vertical (flechas finas). C) La pierna muestra el borde ribeteado híper-intenso entre el músculo soleo y los gastrocnemios (estrellas negras). D,E) Aspecto clínico del paciente con retracciones de codos y cicatriz hipertrofiada (D), e hiperlaxitud llamativa de los dedos(E).
F-K: Características clínicas y patrón radiológico de un paciente de 21 años con una forma intermediaria (Ullrich/Bethlem). F) Inicio de síntomas en la infancia con retraso de marcha e hiperlaxitud, evolución lentamente progresiva con desarrollo de retracciones de codos y tobillos durante la infancia. H-K) La RM muscular de cuerpo entero muestra el aspecto típico en bandas (tigroide), en el que se alternan bandas de híper e hipo intensidad (flechas) en diferentes músculos de diferentes regiones: H) Hombros: bandas radiales en el músculo deltoides (Del), perpendiculares en el músculo subescapular (SC) y supra espinoso (SS). I-J) Imagen de RM del brazo, en secuencia T1-TSE (I) y STIR (J): banda horizontal en el músculo tríceps braquial (TRI). K) Signos de infiltración fibro-adiposa severa y bandas verticales en los músculos paravertebrales lumbares (PV). (Quijano-Roy et al. Congenital Muscular Distrophies, Neuromuscular Imaging - Wattjes & Fischer, Springer, 1st Ed, 2013: 190).
Es una entidad clínica y genética bien definida que se caracteriza a nivel molecular por la falta de selenoproteína N1 secundaria a mutaciones del gen SEPN112. La selenoproteína 1 es una proteína localizada en el retículo endoplasmático. Se desconoce su función exacta pero, parece que participa en la protección de la célula contra el estrés oxidativo. Se postula también que podría afectar a la célula satélite muscular. En contraste con el cuadro clínico (muy homogéneo y reconocible en los pacientes evolucionados), los datos histológicos en la biopsia muscular pueden ser muy diversos y ser informados como distrofia muscular o como diferentes tipos de miopatías congénitas, especialmente de tipo multi-minicore6, con desproporción del tipo de fibras o de tipo desminopatía con cuerpos “Mallory –body like”. El término “selenopatías” a menudo se utiliza para recopilar las diferentes formas relacionadas con mutaciones del gen SEPN1 que comparten el mismo fenotipo. El modo de transmisión es autosómico recesivo.
La presentación clínica es muy estereotipada, con una rigidez selectiva y progresiva de la columna cervico-dorsal y una afectación respiratoria característica durante la primera década de vida11,35. La hipotonía y la debilidad muscular principalmente axial, cervico-dorsal, están presentes desde el primer año de vida, pero no impiden las adquisiciones motrices (que pueden ocurrir a edades normales o estar retrasadas). La marcada inestabilidad y debilidad del cuello y la columna cervico-dorsal persisten durante los primeros años. Posteriormente evolucionan, entre los 3 y los 12 años, a una rigidez progresiva y severa (por la retracción de los músculos extensores para-espinales). Con el tiempo, los pacientes pierden la capacidad de flexionar el cuello y desarrollan una hiperextensión de la nuca. En la visión anteroposterior, es típica una deformación de la columna con translación lateral. En la visión de perfil, existe generalmente una híper-lordosis lumbar asociada a una espalda rectificada, con una penetración de la columna en tórax muy patológica. La disfunción de los músculos respiratorios es prácticamente constante, necesitando ventilación antes de la edad adulta, a pesar de conservar la marcha. Como en los pacientes con DMC de tipo Ullrich, en esta miopatía el diafragma está selectivamente afectado36, con mala tolerancia al decúbito supino y disminución significativa de la capacidad vital en posición acostada con respecto a la posición sentada. La rigidez de columna se acompaña de una rigidez y poco desarrollo de la caja torácica. En ausencia de una ventilación nocturna adecuada, la disfunción respiratoria puede llevar a un fallo multi-orgánico. Los síntomas diurnos pueden ser anodinos e insidiosos. La hipercapnia diurna puede causar dolores de cabeza, trastornos cognitivos, insuficiencia cardíaca secundaria y, si no se trata adecuadamente, el fallecimiento. Esta frecuente y severa complicación implica la necesidad de un seguimiento anual o bianual de la función respiratoria durante el día y de la ventilación nocturna en estos pacientes.
Una atrofia muscular difusa se hace evidente durante la primera década, contrastando con la preservación relativa de la fuerza de miembros. El patrón de afectación muscular es selectivo, con una debilidad y fatigabilidad importante de los músculos más axiales (extensores de cuello y tronco) y de los glúteos y de los aductores del muslo. Los estudios de resonancia magnética han mostrado un patrón muy reconocible, con atrofia del músculo esternocleidomastoideo en el cuello y con infiltración fibroadiposa de los extensores de la nuca, de los paravertebrales, de los glúteos máximos y de varios músculos del muslo (aductor mayor, sartorio y semimembranoso) (Figura 8). Este último músculo es especialmente útil en el diagnóstico diferencial con otras miopatías ya que con frecuencia está muy atrófico o ausente31,37. Con el paso de los años, los pacientes clínicamente desarrollan un cuello fino y alargado y a nivel de los muslos una separación anormal con aspecto de “paréntesis”. Una ligera debilidad facial y una voz nasal también son frecuentes. Las retracciones de miembros están ausentes o son poco importantes y de aparición tardía (codos, tobillos).
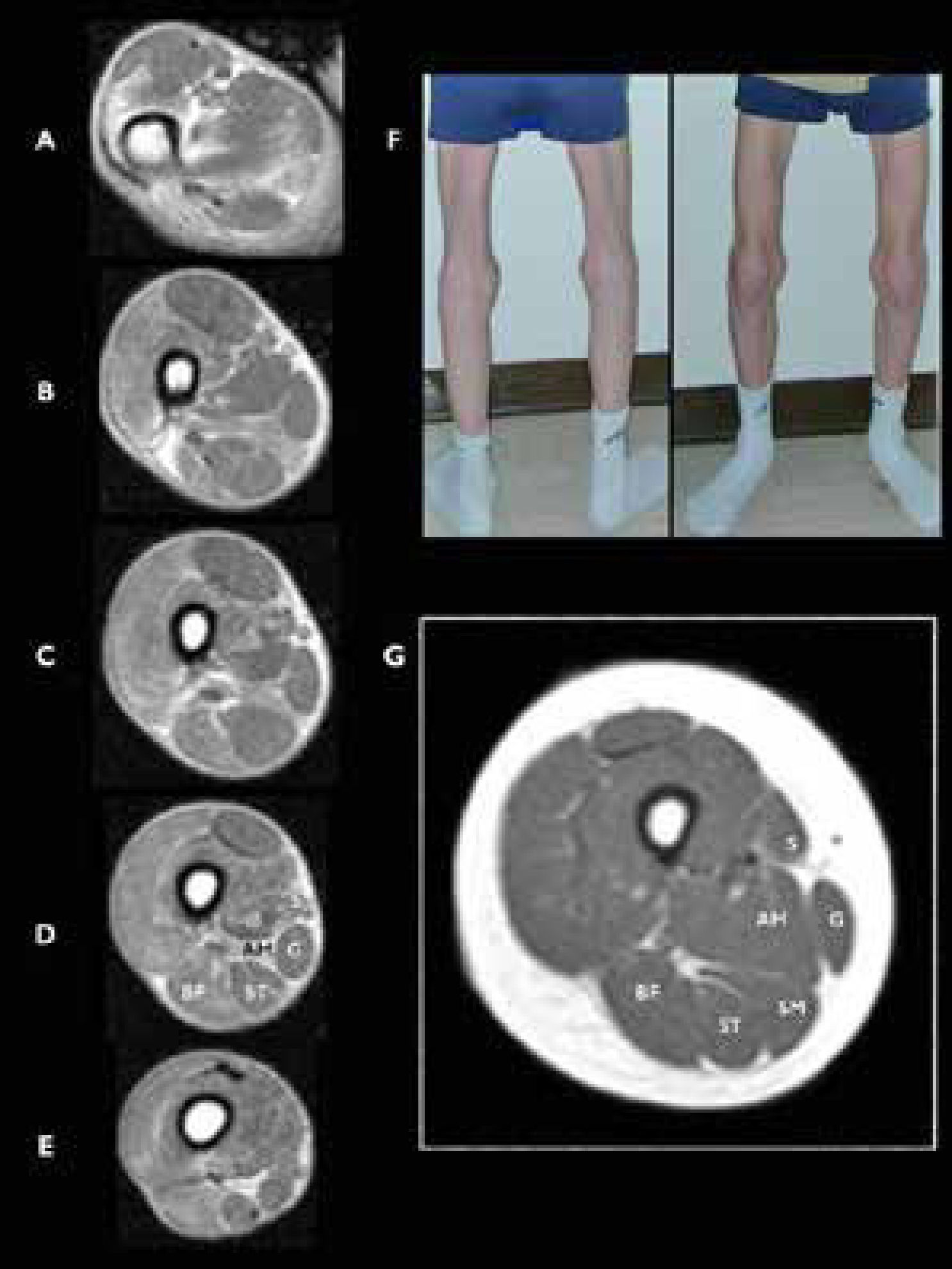
, RM muscular a nivel de muslo A-E) Vista axial a diferentes niveles en el muslo de un joven con rigidez espinal y biopsia distrófica que revelan la ausencia de músculo semimembranoso (SM). F) Imagen de las piernas del paciente explorado en RM, mostrando una atrofia llamativa de los músculos sartorio (vista anteroposterior) y SM (vista posterior). G) Cortes axiales de un muslo de un control sano. Sartorius (S), gracilis (G), adductor magnus (AM), semimembranosus (SM), semitendinosus (ST), y biceps femoris (BF). (Hankiewicz et al. Whole-body muscle magnetic resonance imaging in SEPN1-related myopathy shows a homogeneous and recognizable pattern. Muscle Nerve. 2015 Nov;52(5):733).")
Miopatía relacionada con SEPN1 (SEPN1-RM), RM muscular a nivel de muslo
A-E) Vista axial a diferentes niveles en el muslo de un joven con rigidez espinal y biopsia distrófica que revelan la ausencia de músculo semimembranoso (SM). F) Imagen de las piernas del paciente explorado en RM, mostrando una atrofia llamativa de los músculos sartorio (vista anteroposterior) y SM (vista posterior). G) Cortes axiales de un muslo de un control sano. Sartorius (S), gracilis (G), adductor magnus (AM), semimembranosus (SM), semitendinosus (ST), y biceps femoris (BF). (Hankiewicz et al. Whole-body muscle magnetic resonance imaging in SEPN1-related myopathy shows a homogeneous and recognizable pattern. Muscle Nerve. 2015 Nov;52(5):733).
El diagnóstico precisa una sospecha clínica importante, pues los resultados de biopsia muscular no son específicos. Es más probable encontrar signos distróficos en músculos axiales o proximales, mientras que las biopsias de músculos de las extremidades pueden ser mínimamente “miopáticos”. Debe evocarse en niños mayores o adultos ante la existencia de un retraso de sostén cefálico estable en el primer año o en la aparición más tarde de columna rígida a nivel cérvico-torácico, escoliosis e insuficiencia respiratoria restrictiva grave con parálisis de diafragma en pacientes con buena autonomía motriz6,35. La rigidez espinal no es exclusiva de esta enfermedad y en realidad aparece con el tiempo en todas las DMC, en otras distrofias musculares (Emery-Dreifuss, laminopatías, distrofias de cinturas), miopatías congénitas (titinopatías) e incluso en miopatías metabólicas (enfermedad de Pompe)32. Una particularidad de las selenopatías es que no existen retracciones marcadas a otros niveles excepto en la columna. No hay afectación central o miocardiopatía primaria pero la disfunción cardiaca puede aparecer si la insuficiencia respiratoria no se trata, por lo que hay que controlar la aparición de una hipertensión pulmonar. Las CK plasmáticas son normales habitualmente. La resonancia muscular es muy útil para diferenciarla de las colagenopatías, las laminopatías38, y las miopatías por mutación de gen RYR1, DNM231 y maltasa ácida (enfermedad de Pompe)32,39. La confirmación diagnóstica se realiza mediante el análisis de biología molecular del gen SEPN1, con la identificación de una mutación patógena en cada alelo.
DMC por mutación del gen LMNA (L-CMD)Esta es una forma particular de laminopatía de músculo esquelético descrita en 2008. Se debe a mutaciones en el gen LMNA, que codifica la proteína lámina A/C39. Las laminas son proteínas que desempeñan un papel importante en la arquitectura de la envoltura nuclear por su estructura tridimensional. Además, están implicadas en la organización de la cromatina y la regulación de la transcripción y la replicación de ADN. Las mutaciones en este gen pueden afectar a diversos tejidos conduciendo a diferentes enfermedades, incluyendo patologías del músculo estriado (distrofia muscular de Emery-Dreifuss EDMD2 y EDMD3, distrofia de cinturas tipo LGMD1B), del músculo cardiaco (cardiomiopatía de tipo CMD1A), neuropatías (CMT tipo 2B1), síndromes de lipodistrofia o síndromes de envejecimiento acelerado (progerias). La DMC asociada a mutaciones de la lámina A/C (L-CMD) es la forma más grave y precoz del espectro clínico de laminopatías del músculo esquelético. Mientras que en las laminopatías no congénitas el modo de transmisión puede ser autosómico dominante o recesivo, todos los casos de L-CMD notificados hasta la fecha están vinculados a mutaciones de novo.
La clínica es muy característica, con dos formas de presentación,40,41. Una más grave y precoz, con disminución de movimientos fetales, debilidad e hipotonía al nacimiento y ausencia de desarrollo motor. La otra suele aparecer más tarde, durante el primer o segundo año de vida, con progresos motores a edades muchas veces normales seguido de, una pérdida del sostén cefálico o “cabeza caída” (dropped head sindrome), que es muy característica y que precede un periodo agravación del tono axial y proximal en miembros superiores y distal en miembros inferiores (Figura 3). Posteriormente aparece una rigidez espinal con híper-lordosis de la nuca, tórax y región lumbar que son también muy evocadoras. Los pies se deforman en equino varo y, si la movilidad se reduce, aparecen retracciones severas en las extremidades inferiores (especialmente a nivel de los tendones de Aquiles, rodillas y caderas). A diferencia del EDMD “clásico”, los codos no suelen tener retracciones hasta edades tardías.
En todos los casos se desarrolla una insuficiencia respiratoria restrictiva progresiva con un tórax aplanado. La ventilación asistida es casi siempre necesaria (antes del segundo año de vida en las formas graves neonatales, en las dos primeras décadas en los pacientes con fenotipo de tipo “cabeza caída”). La enfermedad cardíaca suele aparecer antes de la primera década en los pacientes con forma grave precoz y antes de la segunda década en aquellos con fenotipo de cabeza caída. Se trata principalmente de trastornos del ritmo, habiéndose notificado casos de muerte súbita40. Al igual que en otras laminopatías de músculo esquelético, el pronóstico depende de las complicaciones cardiacas (arritmias, cardiomiopatía), necesitando una vigilancia cardiológica estrecha (anual o bianual) con ECG Holter de 24 horas y ecocardiografía, a veces junto con estudios fisiológicos de conducción cardiaca. Es frecuente el tratamiento anti-arrítmico médico o por medio de dispositivos artificiales (pace-maker, desfibrilador)42.
El diagnóstico clínico es fácil de sospechar en las formas con caída cefálica. Si se trata de una forma neonatal severa es menos reconocible y hay que mantener un alto grado de sospecha, pues las pruebas complementarias no son muy específicas. Las CK se elevan de forma variable, pero generalmente no más de 3-4 veces lo normal40. Son niños en general inteligentes. El análisis histológico muestra, en aproximadamente la mitad de los casos, una apariencia distrófica (sobre todo si la biopsia es de músculo deltoides), o modificaciones miopáticas inespecíficas (más frecuente en biopsias de cuádriceps). La inmunohistoquímica de las laminas A/C no es contributiva. La inmuno-detección de merosina y colágeno VI es normal. Puede existir una irregularidad de la expresión de la alfa distroglicano y bandas anormales de la calpaína-3. La RM muscular muestra infiltración fibroadiposa y atrofia difusa con preservación selectiva de los músculos de antebrazo, cuello, cabeza y del psoas en la cintura pélvica. En formas menos evolucionadas, puede haber una afectación selectiva del vasto lateral en el muslo y de sóleo y gastrocnemio medio en la pierna31,32,34,38. Se observa con frecuencia una pobreza de tejido subcutáneo (lipoatrofia).
DistroglicanopatíasLas distroglicanopatías son enfermedades debidas a una anomalía de la glicosilación de la proteína α-distroglicano (α-DG). Una gran heterogeneidad clínica y genética caracteriza a este grupo. Su frecuencia relativa en Europa está en torno al 15%14. La presentación clínica es muy variable, desde síndromes cerebro-musculares con lisencefalia, DMC y distrofias de cinturas con o sin retraso mental, hasta cuadros pseudo-metabólicos con mialgias y rabdomiólisis, sin debilidad muscular significativa (Figuras 3 y 4)43-47. α y β-distroglicano son proteínas altamente glicosiladas codificadas por el mismo gen (DAG1) y se localizan a nivel de la membrana basal del sarcolema de la fibra muscular respectivamente. Desempeñan un papel esencial en la construcción de enlaces entre diferentes proteínas de la membrana celular (distrofina y sarcoglicanos) y de la matriz extracelular a través de lamininas como la merosina. Existen dos tipos de glicosilación O- y N. Mientras que las distroglicanopatías se deben a anomalías en el primer tipo, las anomalías de la N-glicosilación conducen a una vasta familia de enfermedades neurológicas denominadas “síndromes CDG”, que habitualmente no afectan al músculo47. Los distroglicanos también se expresan en el cerebro y el ojo, donde participan en la morfogénesis y la migración neuronal44,45. Todas las mutaciones conocidas hasta la fecha en al menos 18 genes responsables de distroglicanopatías se transmiten de forma autosómica recesiva48. Los primeros identificados, entre 1998 y 2005, fueron el gen de la fukutina (FKTN), responsable de la DMC de tipo Fukuyama (FCMD)2, gen de la beta O-manosa 1,2-N-acetilglucosaminiltransferasa 1 (POMGnT1) identificado en pacientes con enfermedad Muscle-Eye-Brain (MEB)3, gen de la proteína relacionada con la fukutina (FKRP), gen de la O-manosil transferasa 1 (POMT1) y O-manosil transferasa 2 (POMT2), identificados en pacientes con sindrome de Walker-Warburg (WWS), y gen de la proteína Large (LARGE). Pronto fue evidente que no existía una correlación estricta del gen mutado y el sindrome, sino que existe un continuo de fenotipos. Por ejemplo las anomalías en el gen FKRP fueron identificadas en fetos con WWS, en MEB, DMC (MDC1C) y hasta en pacientes inteligentes con mialgias y aumento de CK43-45. Desde 2009 y especialmente en 2012, más de diez nuevos genes han sido identificados como responsables de estas patologías, en pacientes con o sin malformaciones cerebrales, con o sin retraso mental, con o sin neuroimagen normal: ISPD, DPM1, DPM2, DPM3, DOLK, GTDC2, TMEM5, B3GALNT2, B3GNT1, GMPPB, POMK46,47. Además de las distroglicanopatías secundarias, se han identificado mutaciones del propio gen de la aDG (DAG1). Muchos de estos genes codifican glicosil-transferasas (POMT1, POMT2, POMGNT1, LARGE, GTDC2, B4GAT1, B3GALNT2. Recientemente, la función de los genes FKRP, ISPD y FKRP ha sido elucidada49. Las cadenas de carbohidratos contienen estructuras en tándem de ribitol-fosfato y estos tres genes están involucrados en el metabolismo de esta molécula por lo que su ausencia provoca un déficit de ribitol-fosfato48,49.
El diagnóstico puede ser prenatal en fetos con anomalías cerebrales severas. En el periodo post-natal, debe ser evocado clínicamente en lactantes hipotónicos o niños con retraso motor o psicomotor (retraso del lenguaje, trastornos de aprendizaje, autismo, retraso mental) asociado a debilidad muscular cervico-axial y proximal en las extremidades. El aumento marcado de CKs es constante y puede servir para detectar la enfermedad muscular en casos con alteraciones cerebrales están en primer plano. Es frecuente una hipertrofia muscular de miembros inferiores y pueden aparecer macroglosia y miocardiopatía dilatada en el curso de la enfermedad, pudiendo presentar una evolución severa a nivel motor y respiratorio. Los niños que no adquieren la marcha desarrollan un cuadro paralítico y retráctil en la primera década de la vida y una insuficiencia respiratoria progresiva, necesitando ventilación asistida antes de la edad adulta10,18,50. En los casos con adquisición de marcha, el periodo progresivo está retrasado a la segunda década de la vida. El manejo clínico es similar al descrito en otras formas de DMC, necesitando una vigilancia cardiológica anual.
La RM cerebral puede ser normal o mostrar tres tipos principales de anomalías (Figuras 3 y 4): Trastornos de giración cerebral, en particular una lisencefalia de tipo 2 (corteza en empedrado o ‘cobblestone’), anomalías en la sustancia blanca que pueden disminuir con el tiempo (supratentorial)44, malformaciones de la fosa posterior (quistes o atrofia de vermis cerebelosos, hipoplasia pontina)44,45. Estas diferentes malformaciones pueden ser más o menos difusas o focalizadas, aisladas o asociadas entre sí.
Existe una clasificación clínico-radiológica que divide en siete grupos las distroglicanopatías, incluyendo DMCs y distrofias de cinturas43:
- 1.
Sindrome de Walker-Warburg (CMD-WWS): con lisencefalia tipo II (cobblestone o en empedrado) y malformaciones cerebrales, cerebelosas y oculares extremadamente severas, incompatibles con la vida o con una supervivencia muy limitada
- 2.
DMCs de tipo muscle-eye-brain/Fukuyama (CMD MEB/FCMD): malformaciones múltiples del SNC (cerebro, cerebelo o tronco) menos graves que el grupo anterior.
- 3.
DMCs con retraso mental y afectación del SNC estructural localizada (CMD-CRB)
- 4.
DMCs con discapacidad intelectual y sin anomalía morfológica del SNC (CMD-MR), posible microcefalia y/o afectación moderada de la sustancia blanca cerebral
- 5.
DMC sin discapacidad intelectual (CMD-no MR). Incluiría MDC1C.
- 6.
Distrofias de cinturas con discapacidad intelectual (LGMD-MR). Puede incluir microcefalia y/o anormalidades moderadas de la sustancia blanca cerebral.
- 7.
Distrofias musculares de cinturas sin deterioro intelectual (LGMD-no MR). Incluye LGMD2I (FKRP), 2L (ISPD) y 2M (FKTN).
La biopsia muscular permite poner en evidencia la distrofia muscular y realizar análisis inmunohistoquímicos para estudiar la glicosilación de alfa-distroglicano. Las deficiencias parciales pueden ser difíciles de detectar por inmunohistoquímica, siendo la técnica de Western Blot más eficiente, pues permite la evaluación de la cantidad y el tamaño de la proteína. Se puede observar un déficit secundario de merosina.
Los genes más frecuentemente involucrados son POMT1, POMT2, POMGnT1, FKRP e ISPD14,43,45-47. FKTN es el más frecuente en pacientes de origen japonés o asiático en general. Hasta hace pocos años, el diagnóstico en genética molecular sólo era posible en aproximadamente el 50% de los casos de distroglicanopatía con estudio de estos seis genes “clásicos”. La identificación de diez nuevos genes y los estudios de secuenciación a alto débito y paneles han mejorado el rendimiento diagnostico en los últimos años. Existen ciertos marcadores fenotípicos útiles en la práctica clínica: los únicos genes identificados en pacientes con inteligencia conservada son ISPD, FKRP y FKTN. POMGNT1 suele estar asociado a cuadros cerebro-oculares (MEB) donde los pacientes presentan macrocefalia, epilepsia refractaria al tratamiento, retraso mental severo y trastornos del comportamiento. POMT1 y POMT2 pueden estar asociados solamente a un retraso psicomotor con microcefalia, pero no siempre anomalías neuro-radiológicas10,43,46. Muchos de estos genes han sido descritos también en pacientes con formas de distrofias musculares más tardías, distrofias de cinturas, siendo más fáciles de sospechar una distroglicanopatía cuando el déficit muscular está asociado a retraso mental, pues esta asociación no existe en otros tipos de distrofias de cinturas.
CONCLUSIÓNLas distrofias musculares congénitas son un grupo de enfermedades musculares muy heterogéneas en términos clínicos y genéticos. Actualmente hay casi una treintena de genes conocidos responsables, diferenciándose cinco formas principales de DMC (merosinopatías, colagenopatías, distroglicanopatías, selenopatías y laminopatías). El concepto de la distrofia muscular congénita ha evolucionado en los últimos años. Los avances genéticos han demostrado una superposición clínica e histológica con otras miopatías inicio temprano y un continuum de espectros con miopatías más tardías y distrofias musculares de cinturas. El diagnóstico genético puede a menudo ser orientado mediante la identificación de marcadores clínicos (hipermovilidad distal, hipertrofia muscular, contracturas articulares, selectivo de rigidez espinal), la determinación de enzimas musculares, proteínas inmunohistoquímica (merosina, glicosilación de distroglicano, colágeno VI) y anormalidades detectadas por resonancia magnética en el cerebro o el músculo. La identificación y caracterización de estas enfermedades, su historia natural y la mejora del conocimiento fisiopatológico a través del trabajo en los pacientes y de modelos animales permiten esperar la llegada de terapias innovadoras en los próximos años.
Declaración de interesesDeclaro que no tengo conflictos ni financiación en relación con el tema del capítulo escrito.
Declaro que tengo la autorización de los padres y/o pacientes mayores de edad para mostrar las fotografías que estan contenidas en el manuscrito en una revista médica-científica.