




Los conceptos de farmacodinámica deberían aplicarse para optimizar los regímenes de dosificación antibiótica, especialmente de cara a algunas infecciones bacterianas resistentes a múltiples drogas. A pesar de que la farmacodinamia de la mayoría de las clases de antibióticos utilizadas en el ámbito hospitalario está bien descrita, las pautas acerca de cómo seleccionar regímenes y su implementación en un programa de administración antimicrobiana en la propia institución, son más limitadas. La función del antibiótico en concentraciones mínimas inhibitorias (MIC) es primordial para entender qué regímenes podrían beneficiarse de la implementación como un protocolo o como uso en los pacientes individuales. Este artículo destaca la farmacodinámica de aminoglucósidos, betalactámicos, fluoroquinolonas, tigeciclina, vancomicina, polimixinas, con el objetivo de proporcionar una base estratégica para seleccionar un régimen antibiótico optimizado en vuestro ámbito hospitalario.
Las infecciones resistentes a los antibióticos son un problema de salud pública a nivel mundial. Como resultado de una resistencia emergente tanto en bacterias gram-positivas como gram-negativas, los patógenos que permanecen susceptibles a gran parte de los antibióticos actualmente disponibles están disminuyendo y hay pocos antibióticos en desarrollo para hacer frente a estas bacterias resistentes a múltiples drogas (MDR, en inglés)1. Entre las bacterias gram-positivas, los Staphylococcus aureus, que son resistentes a los betalactámicos [por ej., S. aureus resistente a meticilina (MRSA, en inglés)] pueden encontrarse en 50-60% de las cepas aisladas2. Clínicamente hablando, llegamos al punto por el que si S. aureus es una causa sospechosa de infección, la terapia empírica con un antibiótico anti-MRSA se ha hecho esencial. Desde el punto de vista de las bacterias gram-negativas, la Pseudomonas aeruginosa continúa siendo un patógeno problemático debido a su alta prevalencia en el ámbito hospitalario; sin embargo, la emergencia de las enterobacterias resistentes al carbapenem (CRE, en inglés) y de la bacteria Acinetobacter baumannii resistente a carbapenem (CRAB, en inglés) ha llamado mucho la atención puesto que se consideran amenazas urgentes y graves, respectivamente, por los Centros de Control de Enfermedades2,3. La falta de nuevos antibióticos es especialmente problemática en países fuera de Estados Unidos y de la Unión Europea. Muchos de estos países tienen requisitos regulatorios que demoran significativamente la aprobación de nuevas drogas, o en casos extremos, nunca las tienen disponibles. Como resultado, los países que a menudo tienen los niveles directos de resistencia a múltiples drogas (MDR), pocas veces disponen de los antibióticos más potentes y más nuevos en su arsenal.
Además de fomentar el desarrollo continuo de nuevos antibióticos, deben hacerse esfuerzos dentro del ámbito hospitalario para limitar la emergencia y diseminación de bacterias MDR. Los Programas de Administración Antimicrobiana (ASP, en inglés) se han hecho ampliamente populares en los Estados Unidos y en Europa para suplir esta necesidad no satisfecha4. Dichos programas tienen como objetivo controlar y administrar el uso de antimicrobianos en unidades de cuidados intensivos a través de intervenciones coordinadas diseñadas para mejorar y medir el uso apropiado. Por lo tanto, los ASP promueven la selección de regímenes óptimos de fármacos antibióticos así como su dosificación, duración de la terapia y vía de administración en el centro de salud. Uno de los componentes de los ASP es la consideración e implementación de regímenes antibióticos basados en conceptos farmacodinámicos. Aunque el uso de la farmacodinamia para diseñar regímenes de dosificación de antibióticos -tales como la infusión continua de betalactámicos- ha sido ampliamente informado en la literatura, el diseño estratégico e implementación de dichos programas como parte de un ASP no lo ha sido tanto.
En este artículo haremos una breve revisión acerca de cómo se administra la farmacodinamia antimicrobiana, seguido por una discusión de consideraciones y estrategias sobre el mejor lugar donde la implementación de estas estrategias de dosificación puede proporcionar los mejores beneficios.
FARMACODINAMIA: ¿CUÁL ES LA DOSIS CORRECTA?Una terapia antibiótica inapropiada a menudo es resultado de una administración demorada (por ej., esperar los resultados de susceptibilidad o de un cultivo, o antes de iniciar los antibióticos o de comenzar la terapia como resultado de un cultivo positivo) o, más frecuentemente, de una subestimación de las tendencias actuales sobre resistencia. Independientemente de esto, la clasificación de un organismo como “sensible”, “intermedio”, o “resistente” no es suficientemente informativo para determinar la dosis ideal que debe usarse para la infección. En cambio, debiera usarse el término “terapia antibiótica óptima” para indicar no sólo que se ha elegido el antibiótico correcto sino también que la dosis es suficiente para obtener el máximo umbral de exposición determinado por los estudios farmacodinámicos. Una observación relevante para la terapia antibiótica óptima es que el patógeno no necesita ser “sensible” al fármaco en cuestión, mientras la exposición del agente sea suficiente para matar al organismo.
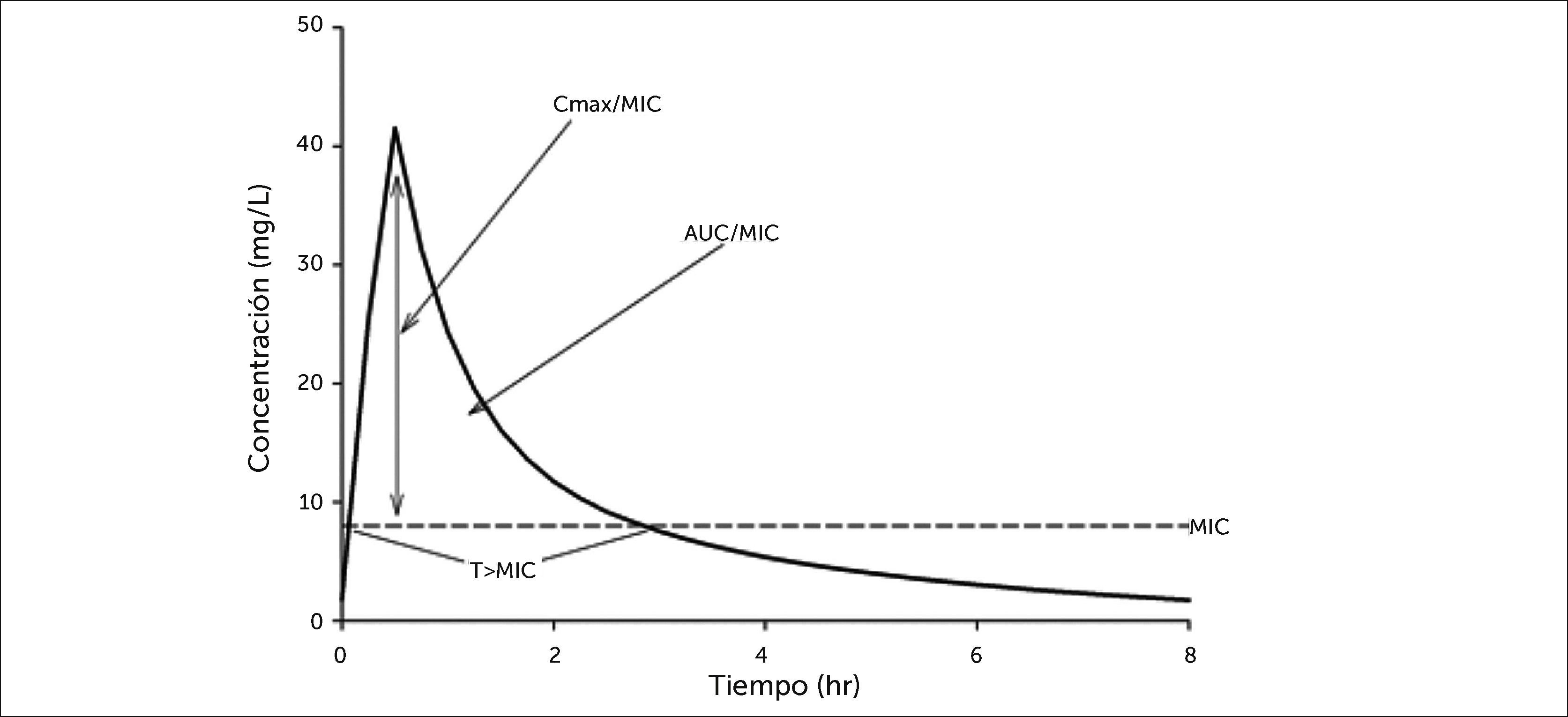
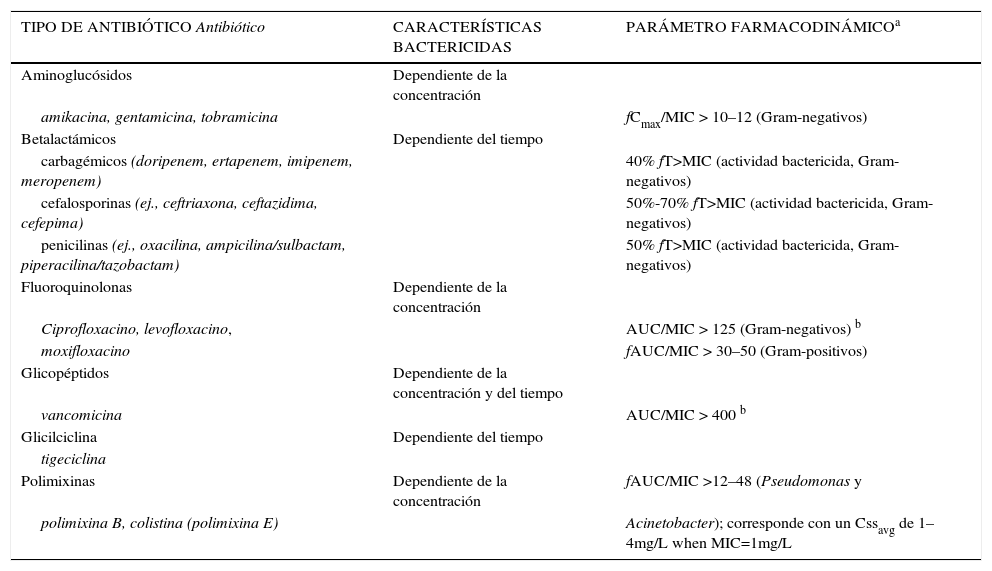
Las características bactericidas antimicrobianas dependen tanto de la concentración del fármaco en relación a la concentración mínima inhibitoria (MIC) como del tiempo durante el cual se mantiene esta exposición (Figura 1)5. Cuando el efecto de la concentración predomina por sobre el del tiempo, el antibiótico despliega efectos dependientes de la concentración que están asociados significativamente a una concentración máxima libre de fármaco y la relación MIC(fCmax/MIC). Cuando el factor tiempo es mayor, el antibiótico tiene efectos dependientes del tiempo y los resultados de las bacterias están asociados a concentraciones libres de fármacos que quedan sobre MIC para una porción definida del intervalo de dosificación (fT>MIC). Además, los antibióticos dependen tanto los efectos de la concentración como de la dependencia del tiempo pueden observar acción bactericida que está asociada con el área bajo la curva libre de droga (fAUC) hacia la relación de MIC (fAUC/MIC). En la Tabla 1 aparece un resumen de tipos de antibióticos disponibles actualmente para su uso en unidades de cuidados intensivos y sus respectivas características farmacodinámicas. En dosis estándares clínicamente relevantes, hay antimicrobianos dependientes de la concentración entre los cuales se incluyen: aminoglucósidos, fluoroquinolonas y colistina. Entre los antimicrobianos dependientes del tiempo se encuentran: los betalactámicos, las glicilciclinas y la vancomicina.

RESUMEN DE ANTIBIÓTICOS QUE DESPLIEGAN CARACTERÍSTICAS BACTERICIDAS DEPENDIENTES DE LA CONCENTRACIÓN O DEL TIEMPO, Y EL REQUISITO DE EXPOSICIÓN FARMACODINÁMICA
| TIPO DE ANTIBIÓTICO Antibiótico | CARACTERÍSTICAS BACTERICIDAS | PARÁMETRO FARMACODINÁMICOa |
|---|---|---|
| Aminoglucósidos | Dependiente de la concentración | |
| amikacina, gentamicina, tobramicina | fCmax/MIC > 10–12 (Gram-negativos) | |
| Betalactámicos | Dependiente del tiempo | |
| carbagémicos (doripenem, ertapenem, imipenem, meropenem) | 40% fT>MIC (actividad bactericida, Gram-negativos) | |
| cefalosporinas (ej., ceftriaxona, ceftazidima, cefepima) | 50%-70% fT>MIC (actividad bactericida, Gram-negativos) | |
| penicilinas (ej., oxacilina, ampicilina/sulbactam, piperacilina/tazobactam) | 50% fT>MIC (actividad bactericida, Gram-negativos) | |
| Fluoroquinolonas | Dependiente de la concentración | |
| Ciprofloxacino, levofloxacino, | AUC/MIC > 125 (Gram-negativos) b | |
| moxifloxacino | fAUC/MIC > 30–50 (Gram-positivos) | |
| Glicopéptidos | Dependiente de la concentración y del tiempo | |
| vancomicina | AUC/MIC > 400 b | |
| Glicilciclina | Dependiente del tiempo | |
| tigeciclina | ||
| Polimixinas | Dependiente de la concentración | fAUC/MIC >12–48 (Pseudomonas y |
| polimixina B, colistina (polimixina E) | Acinetobacter); corresponde con un Cssavg de 1–4mg/L when MIC=1mg/L |
El objetivo de dosificar antimicrobianos dependientes de la concentración es lograr una concentración total de fármacos de Cmax/MIC de aproximadamente 10 a 12 o un total de AUC/MIC de 150, en donde ambos han pronosticado un éxito clínico6,7. Los objetivos de exposición total del fármaco son razonables en este caso porque los tres aminoglucósidos disponibles actualmente (gentamicina, tobramicina, y amikacina) tienen un bajo enlace proteico. Como resultado de los estudios farmacodinámicos, el régimen de dosificación tradicional de 1 a 1.5mg/kg (gentamicina y tobramicina) o 7.5mg/kg (amikacina) divididos en dos a tres dosis diarias ha sido ampliamente reemplazada con altas dosis de intervalo extendido para lograr concentraciones máximas más altas, dando como resultado una mejor eficacia clínica y en forma muy importante, menos eventos nefrotóxicos. Nicolau y colegas evaluaron un algoritmo de dosificación de aminoglucósidos de una vez al día (7mg/kg diarios, conocido como el Nomograma Hartford) en más de 2000 pacientes adultos y encontraron una respuesta clínica similar, pero una reducida incidencia de nefrotoxicidad comparado con los datos históricos (1.2% vs. 3-5%)8. En un estudio de simulación, se determinó la probabilidad de nefrotoxicidad y resolución de la temperatura del día 7 entre un régimen de aminoglucósidos una vez al día (10mg/kg cada 24 horas) y uno con una dosis de dos veces al día (5mg/kg cada 12 horas)9. En una MIC de 4mg/L (punto de interrupción de la susceptibilidad actual de las bacterias gram-negativas), la dosis de dos veces al día tenía un 53.6% de probabilidad de resolución de temperatura comparado con el 79.7% del régimen de una dosis diaria. Además, se pronosticó que la nefrotoxicidad producida por la dosis de dos veces al día iba a ser significativamente mayor (24.6%) que la del régimen de dosis de una sola vez al día (<1%). Por lo tanto, la dosis específica que se necesita para obtener eficacia sería dependiente de la MIC de bacterias gram-negativas en la población clínica y en la función renal del paciente. Si las MICs son menores a 1mg/L, las dosis de 3-5mg/kg de una vez al día serían suficientes para obtener umbrales de exposición adecuados. La dosis de 7mg/kg del Nomograma de Hartford fue diseñada para lograr proporciones óptimas de Cmax/MIC para gentamicina y tobramicina en la MIC de 2mg/L, que correspondía a la MIC90 de P. aeruginosa en la institución en esa época. En contraste MICs de 4mg/L requerirían dosis de 10-14mg/kg diariamente para lograr los objetivos farmacodinámicos necesarios. En el caso de pacientes con función renal normal, estas dosis se administrarían diariamente; sin embargo, en el caso de pacientes con función renal moderada a severa, la re-dosificación debería disminuirse hasta lograr que las concentraciones caigan a menos de 1mg/L. Aunque no haya cambios en las etiquetas de la FDA, la dosis de aminoglucósidos optimizada, alta, de intervalo extendido es, hoy en día, el régimen de dosificación más empleado para este tipo de antibiótico10.
BETALACTÁMICOSLos antibióticos betalactámicos tienen una actividad bactericida dependiente del tiempo y en general, requieren fT>MIC para ∼50% del intervalo de la dosis para lograr los máximos efectos; no obstante, la exposición puede variar según la clase específica del betalactámico. Por ejemplo, se informa que mientras los betalactámicos a base de penicilina requieren 50% de fT>MIC, estudios en humanos y animales con cefalosporinas sugieren un requerimiento entre 50% y 70% de fT>MIC11–13. Generalmente se piensa que los carbapenem (por ej., doripenem, ertapenem, imipenem, meropenem) logran una máxima actividad bactericida a ∼40% fT>MIC14. Como resultado, la estrategia de administración consiste en maximizar el tiempo en que las concentraciones permanecen sobre la MIC. Se han utilizado varios métodos para maximizar T>MIC, entre ellos: dar dosis más altas, administrar los fármacos con mayor frecuencia, y prolongar el tiempo de infusión (ya sea 3-4 horas, dependiendo de la estabilidad de la temperatura ambiente, o en forma continua por 24 horas). En general, la manera más efectiva de optimizar la exposición, especialmente contra las bacterias gram-negativas resistentes a múltiples drogas (MDR), es tanto incrementar la dosis administrada como prolongar la infusión, manteniendo así una concentración sobre las MICs más altas para el tiempo requerido de exposición bactericida. Esto se ha aplicado a los betalactámicos tales como cefepima, doripenem y meropenem en numerosos estudios. En los pacientes con función renal normal, los regímenes de dosificación de 2 gramos cada 8 horas (cada dosis administrada como infusiones prolongadas de 3 a 4 horas) tienen una alta probabilidad de tratar organismos considerados resistente con MICs de 8-16μg/ml y 16-32μg/ml para doripenem/meropenem y cefepima, respectivamente, lo que es significativamente mayor que si el mismo régimen de dosificación fuera introducido durante los 30 minutos estándares15. Los regímenes de dosificación de la piperacilina/tazobactam también pueden optimizarse empleando una infusión continua o prolongada de la infusión. Kim y colegas descubrieron que una dosis de 4.5g cada 6 horas (en donde cada dosis se infunde por más de 3 horas) lograría una exposición farmacodinámica similar a la misma dosis diaria (18.0g) administrada como una infusión continua, y ambas tendrían probabilidades más altas de lograr el objetivo que las dosis de 4.5g cada 6 horas (infusión de 30 minutos)16. Lodise y colegas observaron resultados clínicos superiores luego de implementar un régimen de dosificación de piperacilina/tazobactam en su centro médico donde todas las órdenes de piperacilina/tazobactam para 3.375g cada 6 horas (infusión de 30 minutos) se cambiaron a 3.375g cada 8 horas (infusiones prolongadas de 4 horas)17. En pacientes con infecciones causadas por P. aeruginosa, la infusión prolongada tuvo como consecuencia una tasa de mortalidad de 14 días más baja (12.2% vs. 31.6%, p=0.04) y una estadía hospitalaria más corta (21 días vs 38 días, p=0.02) que alcanzó una significancia estadística cuando estuvo limitada a pacientes en estado crítico con un puntaje APACHE II ≥ 17. Se han realizado una cierta cantidad de estudios clínicos, la mayoría son de diseño observacional con betalactámicos de infusión prolongada o continua. Una revisión más completa de estos estudios está fuera del alcance de este artículo, pero puede encontrarse aquí15,18. Sin embargo, los estudios clínicos diseñados de forma más rigurosa, que comparan la infusión continua directamente al mismo betalactámico administrado como una infusión estándar de 30 minutos, incluye los ensayos I y II de BLING (Grupo de Infusión BetaLactámica), en donde ambos son controlados, aleatorios, multicéntricos, prospectivos y doble ciego19,20. BLING I19 estudió a 60 pacientes con sepsis aguda que fueron aleatorizados para infusiones continuas de piperacilina/tazobactam, meropenem o ticarcilina/clavulanato o las mismas drogas administradas en un horario intermitente. La cura clínica en el grupo que tenía infusión continua fue de un 70% comparado con sólo el 43% (p=0.037) en los pacientes tratados con infusión intermitente. T>MIC también fue significativamente mayor en el grupo de infusión continua. BLING II20 estudió a 432 pacientes procedentes de 25 unidades de cuidados intensivos en Australia, Asia y Europa. Sin embargo, el estudio de más pacientes no encontró una diferencia en la variable principal, que consistió en pacientes vivos sin UCI en el día 28, una variable diferente y más desafiante procedente del primer ensayo. BLING II tenía notables limitaciones incluyendo una alta prevalencia de bacterias sensibles. En resumen, gran parte de los estudios con infusión continua y prolongada de betalactámicos ha mostrado su mayor valor al tratar pacientes que están más gravemente enfermos e infectados con patógenos con MIC más alta (es decir, menos sensibles).
FLUOROQUINOLONASAunque las fluoroquinolonas son consideradas antibióticos dependientes de la concentración, la máxima dosis que puede administrarse en forma segura está limitada por la toxicidad del sistema nervioso central relacionado con la dosis, así una dosis de 10 a 12Cmax/MIC no puede ser efectiva contra muchos patógenos, y el tiempo en que las concentraciones se mantienen sobre la MIC debe considerarse para maximizar la respuesta. Por lo tanto, en muchos estudios farmacodinámicos, el efecto bactericida se ha correlacionado con AUC/MIC21. A menudo se cita una concentración total de AUC/MIC≥125 contra bacterias gram-negativas para un efecto máximo, mientras que las bacterias gram-positivas, como el Streptococcus pneumoniae requieren una concentración libre de AUC/MIC≥3022,23. Sin embargo, es importante considerar qué fluoroquinolona se usó en cada estudio farmacodinámico puesto que el enlace proteico varía sustancialmente entre los agentes y, por lo tanto, los objetivos AUC/MIC pueden ser diferentes. En un estudio de 74 pacientes que recibían ciprofloxacino por graves infecciones nosocomiales predominantemente debido a bacterias gram-negativas, una concentración total de AUC/MIC menor a 125 se asoció con una menor probabilidad de respuesta clínica y microbiológica22. Adicionalmente, concentraciones AUC/MIC sobre 125 y sobre 250 fueron asociadas significativamente con tiempos medios más cortos para la erradicación (AUC/MIC <125: 32 días; 125-250: 6.6 días; >250: 1.9 días; p<0.005). Si se corrige el enlace proteico de ciprofloxacino de 40%, el umbral de fAUC/MIC sería de ∼75. En otro estudio, una exposición ≥87 de levofloxacina fue determinada prospectivamente para predecir la erradicación en 47 pacientes con neumonía nosocomial24. Al corregir el enlace proteico de la levofloxacina, el objetivo de la fAUC/MIC sería ∼65, un valor muy similar a la exposición requerida por la ciprofloxacina contra las bacterias gram-negativas. A pesar de que las fluoroquinolonas son antibióticos ampliamente recetados, desde una perspectiva farmacodinámica, son incapaces de lograr una exposición farmacodinámica óptima en dosis estándares no sólo en bacterias consideradas resistentes sino también para un número de bacterias que el laboratorio de microbiología clasificaría como sensible. Este es el resultado de un valor crítico más alto que aceptable utilizado para definir susceptibilidad en las bacterias gram-negativas (≤1mg/L en el caso de ciprofloxacina y ≥2mg/L en el caso de levofloxacina). Los estudios de simulación farmacodinámica sugieren que los valores críticos adecuados debieran ser 0.25mg/L y 0.5mg/L respectivamente, lo cual debería aumentar significativamente las tasas de resistencia en la mayoría de los hospitales, especialmente contra P. aeruginosa25. Como resultado, incluso los regímenes agresivos como ciprofloxacina de 400mg cada 8 horas y levofloxacina de 750mg cada 24 horas han logrado pocas posibilidades de obtener la exposición farmacodinámica requerida contra las bacterias gram-negativas. El uso empírico de antibióticos como monoterapia para infecciones gram-negativas debiera desalentarse salvo que los datos de la MIC sugieran que una exposición adecuada es factible.
GLICOPÉPTIDOS (VANCOMICINA)Aunque históricamente se ha pensado que el éxito de la vancomicina para curar infecciones causadas por bacterias gram-positivas ha estado asociado con niveles valle, y por ello T>MIC, la información contemporánea sugiere que la proporción AUC/MIC es la que mejor pronostica resultados para este antibiótico dependiente del tiempo26. En estudios de pacientes con infecciones pulmonares causadas por S. aureus se observó que la respuesta de la vancomicina estaba asociada con una AUC/MIC de droga total >345, y que la erradicación microbiológica estaba asociada con una AUC/MIC >400. Datos alternativos que respaldan esto último son proporcionados por estudios que sugieren que las respuestas clínicas en bacteriemia causada por S. aureus eran bajas cuando la MIC era > 1mg/L; en esta MIC, la dosis estándar de vancomicina (1g cada 12 horas) no alcanza estas exposiciones de AUC/MIC en pacientes con función renal normal. Una declaración de consenso proveniente de la Sociedad de Enfermedades Infecciosas (IDSA, en inglés), la Sociedad de Farmacéuticos de Enfermedades Infecciosas (SIDP, en inglés), y la Sociedad Estadounidense de los Farmacéuticos del Sistema de Salud (ASHP, en inglés) recomendó que se administre la dosis de carga de la vancomicina, especialmente en pacientes en estado crítico, seguidas por dosis diarias de 30mg/kg para lograr valles de 15 a 20mg/L26. No obstante, en escenarios clínicos donde la MIC de vancomicina era 2mg/L sin respuesta clínica, se sugirió sólidamente cambiar a otra alternativa antibiótica. El desafío de optimizar vancomicina basada en una proporción AUC/MIC es doble. Primero, estimar una AUC precisa, segundo, se requieren concentraciones múltiples a lo largo de todo el intervalo de dosificación; una estrategia de valle solo o cima sola se necesita para estimar una exposición subestimada de AUC de un 23% y un 14% respectivamente27. Un método que utilice un solo valor de valle, o múltiples concentraciones (por lo menos 2 muestras por sobre el intervalo de dosificación), combinadas con una estimación Bayesiana de la AUC resultó significativamente mejor al predecir la verdadera AUC (∼97% de precisión). El segundo desafío yace en la prueba misma de la MIC. Se permite que haya un error de 100% en una determinación precisa de la MIC en cualquier dirección, significando con ello que una MIC de 1mg/L es la misma que una de 0.5 y 2mg/L, proporcionando de este modo una exposición potencial 4 veces mayor. Un paciente que logra una AUC de 24 horas de 400mg*h/L, infectado con una bacteria reportada como una MIV de 1mg/L puede realmente tener una exposición AUC/MIC entre 200 y 800 basados en la variabilidad de la MIC en sí misma. Como resultado, las pautas orientadoras de IDSA MRSA enfatizan la evaluación de la respuesta del paciente a la terapia28. A pesar de estos desafíos bien documentados, la vancomicina sigue siendo el estándar dorado para el tratamiento de infecciones de MRSA.
GLICILCICLINAS (TIGECICLINA)La tigeciclina es el primer miembro de la clase de antibióticos glicilciclinos y despliega una actividad dependiente del tiempo, su objetivo farmacodinámico más estrechamente asociado con la eficacia es la ƒAUC/MIC. En el modelo de neumonía murina se necesitaron las proporciones de 2.17 y 8.78 de ƒAUC/MIC para producir una reducción logarítmica de 1 y 2 respectivamente contra Acinetobacter spp29. Al utilizar los datos del ensayo clínico Fase 3 en el tratamiento de neumonía adquirida en el hospital, se descubrió que fAUC/MIC ≥0.9 estaba asociada con 8 veces más de posibilidades de éxito clínico30. Luego de una dosis de carga de 100mg seguida por una de 50mg cada 12 horas, la AUC0-24 de la tigeciclina en estado estacionario es ∼4.7mg*h/L. considerando que el enlace proteico de la tigeciclina es 80%, la fAUC0-24 sería de ∼0.94mg*h/L, lo cual es similar a la mediana de fAUC0-24 observada durante el estudio de neumonía adquirida en el hospital, 1.08mg*h/L (rango: 0.356-4.02). Como resultado, las dosis estándares de tigeciclina logran una óptima exposición utilizando el umbral farmacodinámico clínico cuando la MIC es ∼1mg/L, o ∼0.5mg/L si se aplica un objetivo de reducción logarítmica de UFC 1. El valor crítico de susceptibilidad para la FDA (Administración de Alimentación y Fármacos, estadounidense) es ≤2mg/L, mientras que el valor crítico de la EUCAST es ≤1mg/L. Lamentablemente se dispone de información clínica limitada para validar estas observaciones y se ha informado de resultados variables con dosificación estándar de tigeciclina. Un reciente ensayo clínico de 55 pacientes con una bacteriemia producida por A. baumannii y extensivamente resistente a los fármacos, comparó una mortalidad de 14 días entre una combinación de colistina/carbapenem y colistina/tigeciclina31. Los pacientes recibieron una dosis de tigeciclina estándar. La combinación colistina/tigeciclina estaba independientemente asociada con un exceso de mortalidad de 14 días pero sólo en el subgrupo de pacientes con una MIC de tigeciclina mayor a 2mg/L. Debido a los malos resultados clínicos durante los estudios de registro de neumonía, duplicar la dosis de tigeciclina a una dosis de carga de 200mg seguida de una de 110mg cada 12 horas se ha transformado en un tratamiento de moda para tratar bacterias gram-negativas resistentes a múltiples drogas (MDR, en inglés). Esta dosis agresiva mejoró la cura clínica (57.5% vs 30,4%, p=0.05) pero no la mortalidad en unidades de cuidados intensivos (48.4% vs 66.6%, p=0.14) en pacientes en estado crítico con infecciones CRAB y CRE32. En todo caso, la mayoría de los pacientes siguieron recibiendo tigeciclina en combinación con un segundo antibiótico como la colistina.
POLIMIXINASLa polimixina B y colistina (polimixina E) han re-emergido en la práctica clínica debido a su gran actividad gram- negativa contra organismos resistentes a múltiples drogas (MDR). Ambos antibióticos fueron desarrollados durante un tiempo cuando los estudios farmacodinámicos no eran requeridos ni eran ampliamente comprendidos para nuevos compuestos; por lo tanto, hasta hace un tiempo atrás, las recomendaciones de dosificación insertas en la caja eran, en su mayoría, incorrectas. Los regímenes de dosificación contemporánea basados en conceptos farmacodinámicos sólo recientemente han comenzado a ser comprendidos, y gran parte de los datos disponibles han sido contribuidos con colistina. La colistina despliega características bactericidas dependientes de la concentración, y la mayoría de los estudios sugiere que la fAUC/MIC está mejor asociada con actividad bactericida33. Si se utiliza murina, el modelo de infección del muslo, se requiere una fAUC/MIC de 12 para lograr una reducción logarítmica de 2 contra las cepas P. aeruginosa y A. baumannii. Sin embargo en el modelo de infección renal murina, esta exposición de fAUC/MIC aumentó a 48 para una reducción logarítmica de 1; más aún, 2 de 3 cepas de A. baumannii testeadas nunca alcanzaron este nivel bactericida con cualquier nivel de exposición testeada. Considerando que el enlace proteico de la colistina es aproximadamente un 50% en humanos y estimando una exposición mayor a 24 horas, con concentraciones promedio en estado estacionario de 1 y 4mg/L corresponden a proporciones de 12 y 48 fAUC/MIC, respectivamente, cuando la MIC de colistina es 1mg/L. Notablemente, la nefrotoxicidad inducida por la colistina depende de la concentración y aumenta en forma desproporcionada con concentraciones mayores que 2,5mg/L. Por lo tanto, ya debiera ser evidente para el lector que las exposiciones requeridas para una mayor eficacia se traslapan significativamente con aquéllas que producen toxicidad. Más aún, estas exposiciones están a una MIC de sólo 1mg/L; mayores exposiciones proporcionalmente requieren MICs más altas. Al momento de la escritura de este informe, el Instituto de Normas Estándares de Laboratorio Clínico (CLSI, en inglés) y el Comité Europeo sobre Pruebas de Susceptibilidad Antimicrobiana (EUCAST, en inglés) intercambiaban opiniones sobre cómo armonizar los valores críticos de la colistina. EUCAST define susceptibilidad contra P. aeruginosa a ≤4mg/L, y contra A. baumannii y enterobacterias a ≤2mg/L. El CLSI (Instituto de Normas Clínicas y de Laboratorio) utiliza ≤2mg/L para los gram-negativos no fermentables, pero no tiene valores críticos definidos para las enterobacterias. Basada en datos farmacocinéticos contemporáneos de Garonzik y colegas34, la Agencia Europea de Medicamentos (EMA, en inglés) aprobó sugerencias de dosificación actualizadas para pacientes con variados grados de función renal. A éstas siguieron recomendaciones de la Administración de Alimentos y Fármacos estadounidense (FDA). En la Tabla 2 se muestra un resumen de estas nuevas recomendaciones de dosificación. Un estudio de simulación comparó las recomendaciones de dosificación de EMA y FDA con dosificaciones médicas estándares35. Tanto las dosis recomendadas por EMA como por la FDA resultaron ser concentraciones de estado estacionario promedio mayores comparados con las dosis seleccionadas por médicos. La dosificación de EMA proporcionó las concentraciones promedio más altas en los rangos de clearence de creatinina (CrCL). Sin embargo, los regímenes de dosificación recomendados por parte de ambas agencias fueron capaces de proporcionar una alta probabilidad de concentraciones de estado estacionario sobre los 2mg/l cuando la CrCL era ≥80ml/min. Por lo tanto, se aconseja ser cautos al utilizar colistina como monoterapia cuando los pacientes tienen buena función renal, una MIC sobre 1mg/L o ambas.
RECOMENDACIONES DE DOSIS PARA COLISTIMETATO INTRAVENOSO POR RANGO DE CLEARENCE DE CREATININA ACTUALIZADAS POR LA ADMINISTRACIÓN DE ALIMENTOS Y FÁRMACOS ESTADOUNIDENSE (FDA) Y POR LA AGENCIA EUROPEA DE FÁRMACOS (EMA)
| CLEARENCE DE CREATININA (ML/MIN) | FDA EEUU DOSIS DIARIAa | EMA DOSIS DIARIAb |
|---|---|---|
| ≥80 | 2.5-5mg CBA/kg | 9 MIU (∼300mg CBA)c |
| 50 to <80 | 2.5-3.8mg CBA/kg | 9 MIU (∼300mg CBA)c |
| 30 to <50 | 2.5mg CBA/kg | 5.5-7.5 MIU (∼183–250mg CBA) |
| 10 to <30 | 1mg CBA/kg | 4.5-5.5 MIU (∼150-183mg CBA) |
| (or 1.5mg CBA/kg cada 36 horas) | ||
| <10 | NA | 3.5 MIU (∼117mg CBA) |
CBA: Actividad de Colistina Base (1mg de CBA = 2.4mg of colistimetato de sodio= 30.000 IU; cada ampolla de colistimetato de sodio contiene 150mg de CBA); MIU: millones de unidades internacionales.
FDA expresó las dosis en mg/kg de CBA, utilizando peso corporal real excepto individuos obesos, donde la dosificación debiera basarse en peso corporal ideal. Las dosis están divididas en 2-3 dosis diarias. No hay recomendación para dosis de carga.
EMA expresó las dosis en MIU, que se han convertido en mg de CBA para esta tabla. Las dosis están divididas en 2-3 dosis diarias. EMA recomienda una carga de dosis de 9 MIU (∼300mg CBA) en pacientes en estado crítico.
c EMA indica que dosis diarias de hasta 12 MIU (∼400mg de CBA) pueden requerirse para pacientes con buena función renal.
Aunque todavía hay estudios pendientes, se asume que la polimixina B tiene un perfil farmacodinámico similar a la colistina en el sentido de que se requiere ƒAUC/MIC de ∼12 para reducciones logarítmicas de UFC de 233. No obstante, a diferencia de la colistina, la polimixina B no es un profármaco, por lo tanto no se requiere la conversión en una forma activa, y el componente activo del fármaco está disponible en forma inmediata. Subsecuentemente, una dosis de carga de polimixina B debiera alcanzar una concentración de cima activa en forma inmediata. Cuando se usa en combinación con dosis diarias más grandes, la ƒAUC/MIC puede maximizarse más fácilmente. Las recomendaciones de dosificación actuales para polimixina B alcanzan el límite en 1.5 a 2.5mg/kg al día. Sin embargo, un estudio farmacocinético reciente en 24 pacientes demostró que una dosis de carga de 2.5mg/kg como una infusión de 2 horas, seguida de 1.5mg/kg cada 12 horas en infusiones de 1 hora, alcanzarían una AUC diaria total de ∼50mg*h/L en aproximadamente 90% de los pacientes36. Esta exposición sería suficiente para obtener el objetivo de fAUC/MIC de 12 hasta MICs de 2mg/L. Notablemente, el aclaramiento de polimixina B no está afectado significativamente por reducciones en el aclaramiento de la creatinina, de modo que no se necesitan modificaciones tan importantes en la dosificación para esta población. En un estudio retrospectivo realizado por Nelson y colegas37 en pacientes con infecciones en el torrente sanguíneo debido a bacilos gram-negativos resistentes a carbapenem, se observó que el recibo de dosis diarias de polimixina B <1.3mg/kg estaba asociado significativamente con mortalidad de 30 días [OR=1.58; IC95% 1.05 a 1.81; P=0.04]. Más aún, los pacientes con daño renal compensaron en un 82% en comparación a aquéllos que recibieron dosis reducidas de polimixina B.
A pesar de que los datos anteriores con colistina y polimixina B son prometedores para guiar una dosis óptima, la resistencia adaptativa sigue siendo un desafío. Un estudio farmacodinámico in-vitro con varias cepas clínicas de A. baumannii demostraron tener un rebrote significativo de la población total, debido a la emergencia de resistencia adaptativa en todas las cepas38. Esto ocurrió incluso con la presencia de regímenes de dosificación agresiva (es decir, simulando concentraciones promedio en estado estacionario libres de 3mg/L). La resistencia adaptativa a las polimixinas también ha sido descrita con P. aeruginosa y enterobacterias. Como resultado, se fomenta una dosificación óptima de polimixinas, pero administrada sola no tiene respuestas clínicas prometedoras. Por lo tanto, se recomienda una terapia combinada como medida rutinaria.
EL VALOR DE LA MICUn tema común de los conceptos farmacodinámicos revisados anteriormente para todos los antibióticos es la importancia de la MIC. Al determinar un régimen de dosificación optimizado para implementarlo en el escenario hospitalario, la ASP debiera considerar las tendencias de tasas de resistencia locales. Más aún, muchos estudios has enfatizado la importancia de instituir datos específicos. Mientras los modelos de susceptibilidad general pueden identificarse a partir de un antibiograma hospitalario, los detalles sobre las distribuciones de la MIC de organismos con frecuencia están ausentes.
Las verdaderas pruebas de MIC antibióticas no se realizan habitualmente en los laboratorios de microbiología porque implican mucha mano de obra y son más costosas que las pruebas automatizadas de susceptibilidad (Vitek II™, Microscan™, etc). Además, la mayoría de los prescriptores no han recibido la capacitación para interpretar adecuadamente la MIC. Por estas razones, el laboratorio de microbiología generalmente sólo conduce pruebas de valores críticos, que es sinónimo de pruebas de MIC pero sólo sobre un pequeño rango de diluciones en torno a valores críticos de susceptibilidad y resistencia. Por ejemplo, si los valores críticos de la susceptibilidad y resistencia de un antibiótico son ≤8mg/L y ≥32mg/L respectivamente, la mayoría de los sistemas automatizados sólo va a testear estas concentraciones. Por ejemplo, si las bacterias no crecen a 8mg/L, se reporta “susceptibilidad”. Sin embargo, no se puede determinar si la MIC es 8mg/L (es decir, sensible a borderline) o mucho más bajo (por ej.; 0.5mg/L). De la misma manera, si el organismo crece en ambas concentraciones (8 y 32mg/L), entonces es reportado como resistente, pero claramente el organismo puede tener una MIC de 32mg/L y ser tratada potencialmente con una dosis más alta de antibiótico que una dosis estándar, o la MIC puede ser mucho más alta (ejemplo, 256mg/L), en cuyo caso, no será posible obtener la exposición bacteriana requerida sin aumentar significativamente el riesgo de toxicidad del paciente. Los datos de la MIC son muy útiles al momento de considerar la farmacodinámica antibiótica puesto que la exposición del fármaco siempre está referenciado a la MIC cuando se trata de decidir cuánta y sobre qué intervalo de dosificación se administrará un antibiótico.
Las pruebas de MIC pueden conducirse usando varios métodos: microdilución en caldo; macrodilución; dilución en agar; Etest® (BioMérieux, Durham, NC, USA); un tipo de test de difusión que utiliza tecnología de gradiente; y finalmente con algunos sistemas automatizados. El Sistema de Microbiología Automatizado BD Phoenix™ (BD Diagnostics, Sparks, MD, USA) y Vitek® 2 (BioMérieux, Durham, NC, USA) también proporcionarán resultados de MIC para una combinación antibiótico/bacterias, pero sólo sobre unos pocos rangos de dilución. Por ejemplo, las MICs de cefepime para gram-negativas en el test del sistema BD Phoenix ™ desde 0.5 a 16mg/L que nuevamente no informa al proveedor si un organismo es potencialmente objeto de tratamiento con una infusión de dosis prolongada más alta que 32mg/L. Cuando sea factible, será preferible el uso de microdilución en caldo o Etest a recopilar datos en MICs distribuidas localmente (por hospital o por unidad); como también podrá utilizarse en pacientes individuales afectados por infecciones resistentes a múltiples fármacos (MDR) para ayudar a optimizar la terapia antibiótica, puesto que ambas metodologías proporcionarán un rango de MICs más amplio para ser testeado.
CÓMO IMPLEMENTAR REGÍMENES OPTIMIZADOS POR PATRONES DE SUSCEPTIBILIDAD ANTIBIÓTICA (ASP, EN INGLÉS)Los ASP pueden considerar 2 métodos diferentes a la hora de optimizar el uso de un antibiótico. El tradicional consiste en enfocarse en el antibiótico en sí: cada vez que se recete un antibiótico, éste será optimizado para el paciente individual. El segundo consiste en enfocar el tratamiento de la infección en sí utilizando la estrategia más óptima. Con respecto a la implementación de un régimen de dosificación antibiótica optimizada en la institución, la segunda estrategia tiene más mérito. Antes de determinar a qué antibiótico y qué régimen de dosificación aplicarle optimización, es crucial entender cuáles son los patógenos más probables que causan la infección (por ej., neumonía asociada al respirador) y las MICs para estas bacterias que causan las infecciones.
El camino clínico de la neumonía asociada el ventilador en nuestro hospital, fue instituido luego de la recolección durante 8 meses de datos de vigilancia de las bacterias y pruebas de MIC39. Los modelos farmacodinámicos fueron empleados sobre la base del agente causal más frecuente para lo cual se dispuso de los datos de la MIC, P. aeruginosa, para determinar la elección del antibiótico y régimen de dosificación de manera tal de proporcionar la mayor probabilidad de obtener su exposición farmacodinámica bactericida. Ambos regímenes de infusión continua y prolongada así como dosificaciones estándares fueron evaluados contra la población de P. aeruginosa. Debido a la creciente resistencia en ciertas UCIs de nuestro hospital, se requirieron regímenes de infusión prolongada de altas dosis de cefepima o meropenem (2g cada 8 horas durante infusiones de 3 horas) para lograr una exposición óptima, puesto que era altamente probable que estos regímenes lograran una exposición farmacodinámica contra cepas con MICs de hasta 32 y 17mg/L, respectivamente. Además, la tobramicina de 7mg/kg una vez al día fue defendida debido a la frecuencia de organismos resistentes a múltiples drogas y a la MIC90 para la población de P. aeruginosa que permanecía en 2mg/L. Se dejó de indicar fuertemente el tratamiento con fluoroquinolonas y se reservó para pacientes que no podían conseguir aminoglucósidos. Finalmente se inició un protocolo de vancomicina de dosis alta apuntando hacia valores valle en el rango de 15-20μg/ml para cubrir el SARM (Staphylococcus aureus resistente a meticilina). Luego de iniciado el protocolo, aprendimos que nuestra población de SARM tenía, en forma predominante, MICs de vancomicina de 1.5 a 2mg/L. Como resultado, ahora permitimos que el prescriptor cambie de terapia a linezolid si un paciente con SARM no presenta mejoría en el día 3 de terapia con vancomicina de dosis alta. Estos regímenes de dosificación fueron protocolizados en las UCIs utilizando un sistema informático de órdenes médicas. Se proporcionó capacitación para todos los prescriptores, enfermeras(os), y farmacéuticos sobre el antecedente/justificación del programa y cuándo usarlo.
Luego de 12 meses de uso, se recopiló información para evaluar el impacto (tanto resultados clínicos como de conformidad) de la vía clínica. Hubo una conformidad cercana al 100% y 94 pacientes fueron tratados por neumonía asociada a respirador durante ese tiempo. Comparados con los 74 pacientes usados como controles históricos, los pacientes tratados por la vía clínica con regímenes de dosificación optimizados con cefepima o meropenem tuvieron menos mortalidad relacionada a infecciones (8.5% vs 21.6%, p=0.029), estaban más propensos a recibir un antibiótico con actividad contra el agente patógeno empíricamente (71.6%, vs 48.6%, p=0.007), tenían menos resistencia a múltiples fármacos (9.6% vs 27.0%, p=0.006) y menos infección relacionada con la extensión de estadía hospitalaria (10.5 vs 23 días, p<0.001). En un análisis económico se observó ahorros aproximados de US$ 40000 por paciente tratado por la vía médica40. Este programa todavía es un protocolo obligatorio en nuestras UCIs, a pesar de que aún hacemos modificaciones a nuestras antibióticos y los regímenes de dosificación luego de investigar las MICs cada dos años. Más recientemente, un régimen de piperacilina/tazobactam de infusión prolongada (4.5g 6h como una infusión de 3 horas) se ha implementado en forma transversal en nuestro sistema de salud basado en datos de MIC, farmacocinéticos contemporáneos, y el uso de bombas inteligentes en el sistema.
En el caso de la vía clínica arriba mencionada, la implementación sólo fue en las UCIs, lo que facilitó la educación y monitoreo. También focalizamos nuestra estrategia de optimización en torno a los betalactámicos, aminoglucósidos y vancomicina, puesto que estos antibióticos eran más apropiados para los patógenos causantes observados en neumonía asociada con respirador. Afortunadamente, agentes tales como polimixina B y tigeciclina son raramente requeridos en nuestro hospital debido a pocos organismos CRAB y CRE. Sin embargo, si esto es diferente en otro hospital, la selección de dosis y su implementación debiera seguir la misma estrategia descrita arriba, la cual debiera incluir en primer lugar, una comprensión de las distribuciones de la MIC para su población, seguida por la implementación del régimen de dosificación más óptimo para cubrir la mayoría de estos patógenos. Una evaluación de seguimiento después de un período de tiempo definido (o número de casos tratados) es esencial para asegurar conformidad (compliance) y que los resultados estén en línea con las expectativas. Con todo, un error crucial pero común sería simplemente implementar un régimen de dosificación optimizada que haya sido descrito en la literatura o utilizada en otro hospital sin considerar la epidemiología local, ya que los resultados pueden variar enormemente.
RESUMENEl continuo aumento de bacterias resistentes a múltiples fármacos (MDR, en inglés) en el ámbito hospitalario ha fomentado la necesidad de patrones de susceptibilidad antibiótica (ASP) y el uso de regímenes de dosificación optimizados desde el punto de vista farmacodinámico que aseguren un tratamiento agresivo de estas infecciones. Entre los regímenes de dosificación optimizados para los betalactámicos está combinar dosis más altas con infusiones continuas o prolongadas para aumentar la fT>MIC. Por contraste, los aminoglucósidos requirieron el uso de dosis más altas y de intervalos extendidos. Finalmente, los regímenes de tigeciclina y polimixina B también requirieron dosis más altas combinadas con intervalos de dosificación similares para aumentar la probabilidad de alcanzar exposición farmacodinámica. Sin embargo, la implementación exitosa de uno de estos regímenes requiere una comprensión completa de la epidemiología local y de la MIC antes de seleccionar algún régimen.
El autor declara no tener conflictos de interés, en relación a este artículo.