



Hereditary neuropathies are the most common inherited neuromuscular diseases. The most prevalent group of these disorders is that of hereditary sensory-motor neuropathies, better known by the eponym of Charcot-Marie-Tooth disease (CMT). These neuropathies present with distal, symmetric and progressive involvement and are associated with point mutations or variations in the number of copies of genes responsible for the coding of proteins with strategic functions in Schwann cells or in axonal structures. To date with advances of the next generation sequencing, more than 80 genes have been identified. Although there is currently no curative treatment to CMT, pharmacological strategies and genetic therapies are under study aiming to modulating the altered expression of some of the main causative genes.
Las neuropatías hereditarias son las enfermedades neuromusculares hereditarias más comunes. El grupo más prevalente de estos trastornos es el de las neuropatías sensoriomotoras hereditarias, mejor conocidas por el epónimo de enfermedad de Charcot-Marie-Tooth (CMT). Estas neuropatías se presentan con afectación distal, simétrica y progresiva, y están asociadas a mutaciones puntuales o variaciones en el número de copias de genes responsables de la codificación de proteínas con funciones estratégicas en células de Schwann o en estructuras axonales. Hasta la fecha, con los avances de la secuenciación de la próxima generación, se han identificado más de 80 genes. Aunque actualmente no existe un tratamiento curativo para la CMT, se están estudiando estrategias farmacológicas y terapias genéticas con el objetivo de modular la expresión alterada de algunos de los principales genes causantes.
Hereditary neuropathies comprise a heterogeneous group of genetic diseases characterized by peripheral nervous system (PNS) impairment, most often with distal, symmetric and progressive involvement. The symptoms classically begin in the first two decades of life, with involvement of the feet, and weakness in the upper limbs can occur in the evolution of the disease.
The most prevalent group of these forms of disease is that of hereditary sensory-motor neuropathies, better known by the eponym of Charcot-Marie-Tooth disease (CMT). The various forms of CMT are classified according to nerve conduction velocity, the predominant type of nerve damage (demyelinating or axonal), inheritance pattern, age of onset of symptoms and specific genetic mutations.
Previously to the original description of the disease, reports of fibular muscular atrophy had already been performed by Virchow, Friedreich and Osler. However, it was only in 1886 that the French neurologist Jean-Martin Charcot and his assistant, Pierre Marie, published the description of five cases of what they called progressive muscle atrophy, assuming the lesion was primarily in spinal cord. Three years later, the Englishman Howard Henry Tooth, for his graduation at Cambridge University, presented the thesis on the fibular form of progressive muscular atrophy, locating the pathophysiological alteration of the disease in the peripheral nerve. However, the current classification related to the pathological characteristics only began from 1968 with the studies of Peter J. Dyck and Edward Lambert1. Despite its old description, understanding about CMT has expanded, as new genes and new pathophysiologic mechanisms have been discovered. This allows the creation of future perspectives for treatment that may modify the natural history of the disease.
ETIOLOGY AND PATHOGENESISCMT and other hereditary neuropathies represent a group of diseases associated with point mutations or variations in the number of copies of genes responsible for the coding of proteins with strategic functions in Schwann cells or in axonal structures2,3. These proteins exhibit physiological and developmental roles that include myelin formation, participation in mitochondrial function, retention in the endoplasmic reticulum, RNA processing, fission and fusion of membranes, and acting as transcription factors and cytoskeletal components. Currently, more than 80 genes are related to CMT and other associated hereditary neuropathies, a number that has shown rapid growth in recent years due to the increasing incorporation of new generation sequencing technology5.
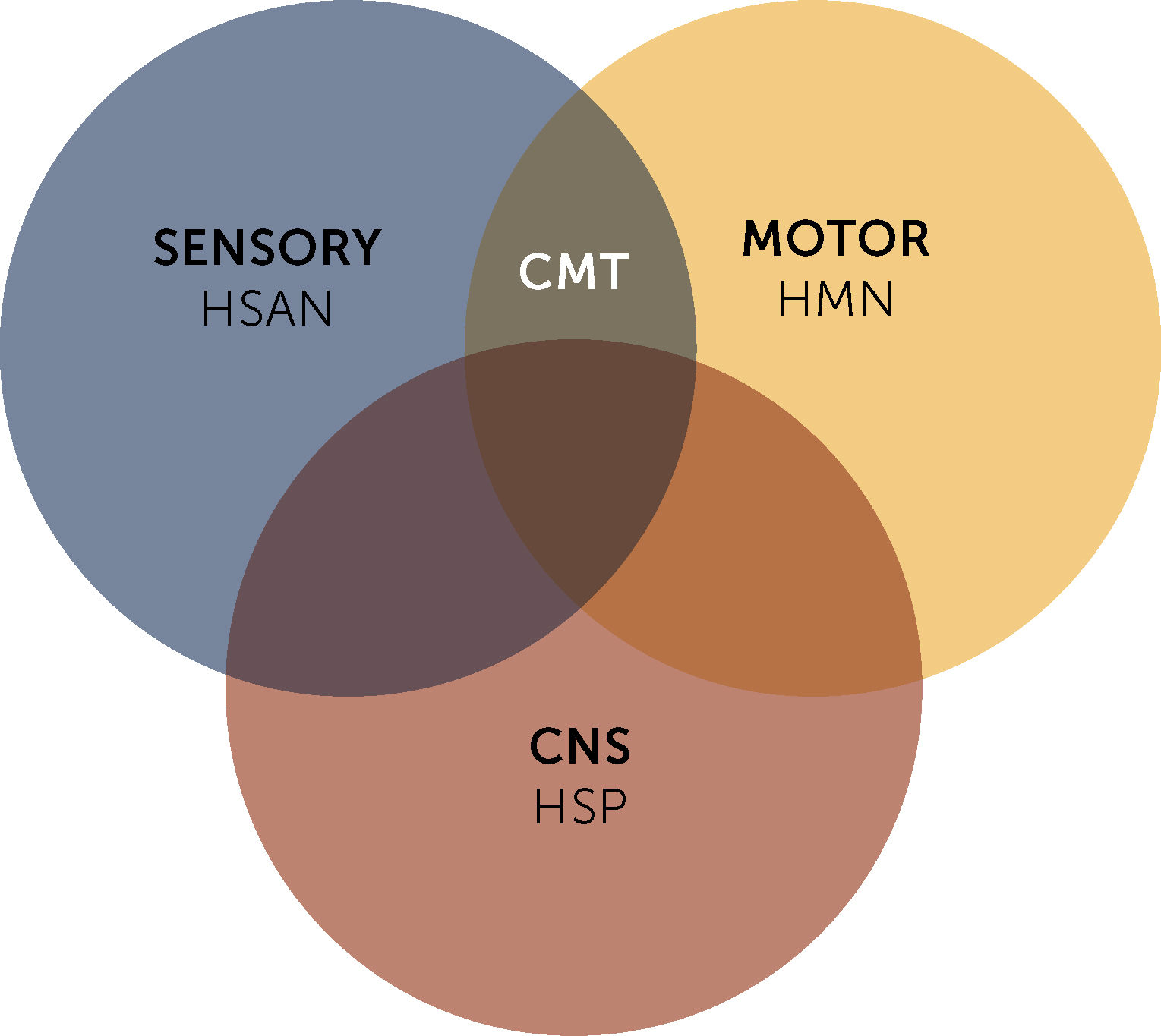
Eventually, CMT is associated with other groups of rare hereditary neuropathies, such as hereditary motor neuropathies (HMN), which present selective impairment of the inferior motor neuron and motor fibers, and hereditary sensory and autonomic neuropathies (HSAN). As well as other diseases that present selective or combined central nervous system involvement, such as hereditary spastic paraparesis (HSP) (Figure 1)2.
 Hereditary sensory and autonomic neuropathy (HSAN) and hereditary motor neuronopathies (HMN) - and hereditary spastic paraparesis (HSP). CNS, Central nervous system.")
The molecular and genetic aspects of CMT are complex and reveal some intriguing interrelations. As the most common form of CMT, CMT1A is caused by duplications in the PMP22 gene responsible for coding the protein that corresponds to about 2 to 5% of the myelin of the PNS. It is produced primarily by Schwann cells and is expressed in the compact portion of myelin in the PNS. On the other hand, deletions in the same PMP22 are related to the hereditary neuropathy with a liability to pressure palsy (HNPP)6.
CMT1B is related to mutations in the gene responsible for the coding of MPZ, which represents up to 50% of the structural protein in the myelin in the PNS. Interestingly, mutations isolated in MPZ gene (which is only expressed in Schwann cells) may also lead to axonal forms of CMT (CMT2I and CMT2J), indicating that the structural interaction between the myelin sheath and the axon is crucial for the maintenance of the axonal integrity7.
The axonal forms of CMT are mainly represented by CMT2A2. The mutated gene in this presentation is MFN2, which encodes mitofusin-2. This protein is located in the outer layer of the mitochondria and interacts with the Miro-Milton complex, which functions in the anchorage of the mitochondria with the motor proteins of the kinesins family8.
Mutations in the GJB1 gene are responsible for CMTX1, which corresponds to the most prevalent form of X-linked CMT and it is also the second most common CMT in the general population. The product of this gene (the gap junction protein, beta-1, also known as connexin 32) has the function of connecting with the different folds in the cytoplasm of Schwann cells, allowing the transfer of nutrients, ions and molecules to the layers internal myelin9.
EPIDEMIOLOGYCMT corresponds to the most common group of inherited conditions within neuromuscular diseases, with a general prevalence of 1 in every 2,500 individuals10. This number may vary regionally and according to different ethnic groups, as well as the types of CMT found. Like the Occidental countries, where there is greater population diversity, dominant and X-linked forms are the most commonly found. However, in more isolated countries with a more homogeneous population, consanguineous marriages are more common, favoring the occurrence of recessive types of CMT3.
In recent years, several epidemiological studies have described the prevalence of specific forms of CMT in specialized centers and in the general population. CMT1 variants represent about half of the diagnosed cases, with mutations in the PMP22 (CMT1A), MPZ (CMT1B), GJB1 (CMTX1) and MFN2 (CMT2A2) genes responsible for 90% of the molecular confirmed forms. All other CMT-related genes account for less than 1% to 2% of individual diagnoses. However, in a percentage of individuals studied, molecular confirmation is not achieved, demonstrating that other genes that cause CMT still require identification3.
GENERAL CLINICAL ASPECTSCMT type 1 (CMT1) refers to the sensory-motor hereditary neuropathies of a demyelinating character, while the axonal forms are classified as CMT2. Both forms have an autosomal dominant inheritance pattern (with few exceptions), and usually begin in childhood or early adulthood. However, later presentations may occur, especially in cases of CMT211.
Dejerine-Sottas disease (previously determined as CMT3) is a form of autosomal dominant or recessive neuropathy of early onset, usually before five years of age. It is characterized by severe demyelination or hypomyelination12.
CMT4 is a sensory-motor neuropathy with an autosomal recessive inheritance that typically begins in childhood or early adulthood11.
The intermediate types of CMT are represented by neuropathies that present different electrophysiological characteristics (demyelinating or axonal) in affected individuals of the same family. Nerve biopsy shows mixed findings of demyelination and axonal damage. The dominant autosomal forms are the most common (CMTDI), but autosomal recessive forms (CMTRI) are also described13.
CMT forms attached to the X chromosome are common in the group of hereditary neuropathies. They should be suspected in families where there is no clear evidence of father-to-child transmission. Some of its subtypes have a dominant X-linked pattern, with carrier women presenting milder phenotypes in some cases9.
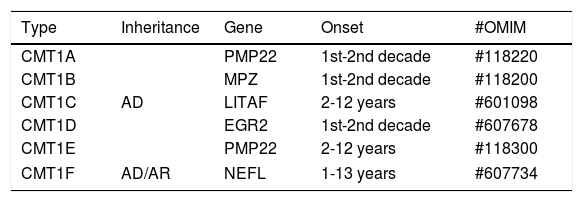
CHARCOT-MARIE-TOOTH DISEASE TYPE 1CMT1 is the most common form of hereditary neuropathy, with the proportion of CMT1: CMT2 being close to 2:1. At least six different subtypes of CMT1 are recognized (Table 1). Typically, CMT1 patients initiate distal lower limb weakness in the first to third decade of life. The anterior compartment is the first to be affected, with involvement of the fibular muscles. This pattern of weakness leads to the development of progressive “foot drop”, with frequent occurrence of stumbling, falls and ankle sprains. Sensitive complaints may be present, but when these symptoms are prominent the diagnosis of acquired neuropathy should be considered14.
Classification of CMT type 1.
| Type | Inheritance | Gene | Onset | #OMIM |
|---|---|---|---|---|
| CMT1A | AD | PMP22 | 1st-2nd decade | #118220 |
| CMT1B | MPZ | 1st-2nd decade | #118200 | |
| CMT1C | LITAF | 2-12 years | #601098 | |
| CMT1D | EGR2 | 1st-2nd decade | #607678 | |
| CMT1E | PMP22 | 2-12 years | #118300 | |
| CMT1F | AD/AR | NEFL | 1-13 years | #607734 |
AD: autosomal dominant, AR: autosomal recessive.
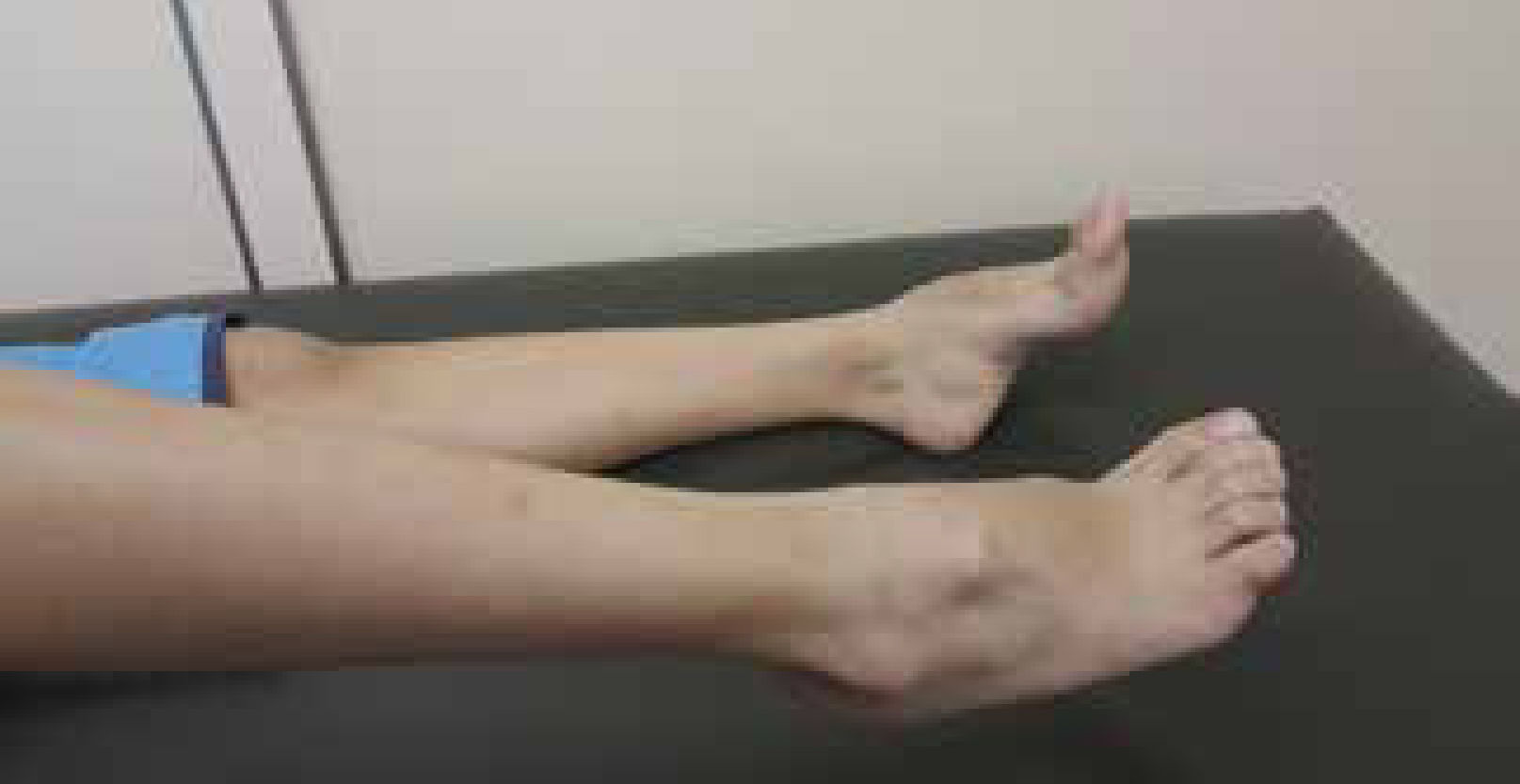
Although individuals with CMT1 generally do not complain of altered sensation in the lower limbs, reduction of sensation in all modalities is evident on clinical examination. In addition, pain and symptoms derived from loss of small diameter afferent fibers is not uncommon in CMT1A15. Osteotendinous reflexes are hypoactive or absent. Usually the distal musculature of the leg is atrophied (leading to the “inverted champagne bottle” aspect). Asymmetric calf pseudohypertrophy may occur rarely. Deformity of the feet is the usual, and there may be development of cavus foot, equinovarus or “hammer toes” (Figure 2). Mild to moderate weakness in the proximal limb region may also happen over time. Involvement of the phrenic nerve, with respiratory compromise, is possible in some cases14.
.")
The involvement of the upper limbs occurs in up to two-thirds of the individuals, with distal weakness and atrophy of the arms, with development of “claw-hands” deformity in the most severe cases. Tremor in hands can be observed in some patients.
Hypertrophy of peripheral nerves, especially in the posterior region of the ears and arms, can be visualized and palpated14.
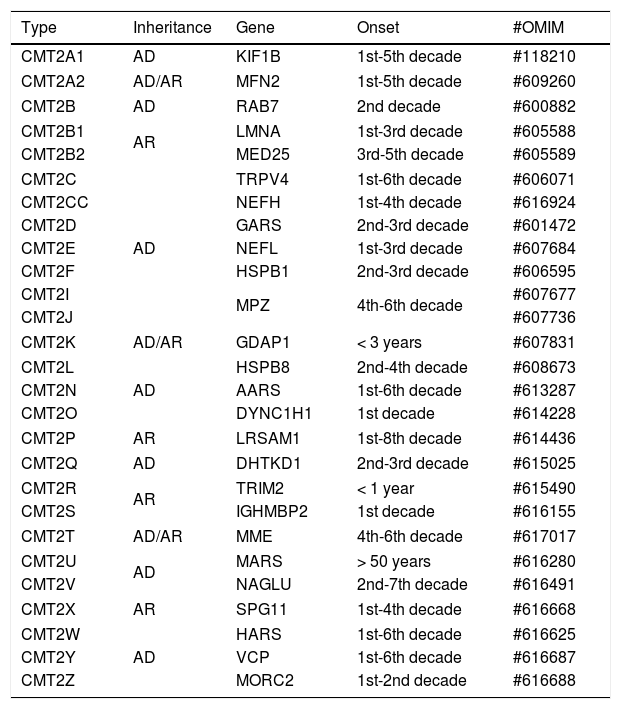
CHARCOT-MARIE-TOOTH DISEASE TYPE 2CMT2 refers to hereditary sensory-motor neuropathies that present electrophysiological findings compatible with primarily axonal involvement. This group of diseases accounts for about 25 to 30% of all cases of CMT, but molecular diagnosis is only done in about a half of patients, even in large centers16.
Most of these forms of disease are associated with an autosomal dominant inheritance pattern, but there are reports of mutated cases in homozygosis or composite heterozygosis, even in its most common subtype, CMT2A217, as well as in rare forms (for example: CMT2B1, CMT2H, CMT2K, CMT2R and CMT2S) (Table 2).
Classification of CMT type 2.
| Type | Inheritance | Gene | Onset | #OMIM |
|---|---|---|---|---|
| CMT2A1 | AD | KIF1B | 1st-5th decade | #118210 |
| CMT2A2 | AD/AR | MFN2 | 1st-5th decade | #609260 |
| CMT2B | AD | RAB7 | 2nd decade | #600882 |
| CMT2B1 | AR | LMNA | 1st-3rd decade | #605588 |
| CMT2B2 | MED25 | 3rd-5th decade | #605589 | |
| CMT2C | AD | TRPV4 | 1st-6th decade | #606071 |
| CMT2CC | NEFH | 1st-4th decade | #616924 | |
| CMT2D | GARS | 2nd-3rd decade | #601472 | |
| CMT2E | NEFL | 1st-3rd decade | #607684 | |
| CMT2F | HSPB1 | 2nd-3rd decade | #606595 | |
| CMT2I | MPZ | 4th-6th decade | #607677 | |
| CMT2J | #607736 | |||
| CMT2K | AD/AR | GDAP1 | < 3 years | #607831 |
| CMT2L | AD | HSPB8 | 2nd-4th decade | #608673 |
| CMT2N | AARS | 1st-6th decade | #613287 | |
| CMT2O | DYNC1H1 | 1st decade | #614228 | |
| CMT2P | AR | LRSAM1 | 1st-8th decade | #614436 |
| CMT2Q | AD | DHTKD1 | 2nd-3rd decade | #615025 |
| CMT2R | AR | TRIM2 | < 1 year | #615490 |
| CMT2S | IGHMBP2 | 1st decade | #616155 | |
| CMT2T | AD/AR | MME | 4th-6th decade | #617017 |
| CMT2U | AD | MARS | > 50 years | #616280 |
| CMT2V | NAGLU | 2nd-7th decade | #616491 | |
| CMT2X | AR | SPG11 | 1st-4th decade | #616668 |
| CMT2W | AD | HARS | 1st-6th decade | #616625 |
| CMT2Y | VCP | 1st-6th decade | #616687 | |
| CMT2Z | MORC2 | 1st-2nd decade | #616688 | |
AD: autosomal dominant, AR: autosomal recessive.
As stated, CMT2A2 is the largest representative of this group and accounts for about one-third of CMT2 cases. It is mostly presented with the classic phenotype of CMT, but some characteristics may help in differentiation with the forms of CMT1. In CMT2, the symptoms usually start later, usually in the second decade, and the involvement of the distal musculature of the upper limbs is less pronounced, with deeper tendon reflexes in the arms usually preserved. In contrast, distal lower limb muscle atrophy is more severe in CMT2 (in CMT2A2, most patients require a wheelchair by age 20). The association with tremors is less observed when compared to patients with CMT1. Deformities like cavus foot or “hammer toes” may be observed, however, at a lower frequency than in CMT114,18.
Although most patients do not complain of sensory loss or paresthesia, 50 to 70% present evidence of reduced sensation for most sensory modalities. In some situations, there may also be hearing loss and optic atrophy. Rarely, in some cases, there is the development of pyramidal signs, with hyperreflexia and hypertonia18.
DEJERINE-SOTTAS DISEASEInitially described in 1893, Dejerine-Sottas disease (DSD) refers to a hereditary sensory-motor polyneuropathy of very early onset, usually before the age of five. The use of the eponym is preferable rather than the early determination of CMT3 to describe children who are severely and precociously affected, independent of the specific mutation3. The inheritance pattern is heterogeneous, and in most cases spontaneous mutations occur in heterozygosis in the PMP22, MPZ or ERG2 genes (the same genes responsible for most cases of CMT1). Mutations of the PRX gene on chromosome 19 are also observed19.
The forms that present at birth with hypotonia, delayed motor development, respiratory insufficiency and difficulty in sucking can be classified as congenital hypomielinizing neuropathy. Usually these cases present with important distal contractures (congenital multiple arthrogryposis), which can be diagnosed even in prenatal examinations. In classical forms, the lower limbs are affected before the upper limbs. There may be development of kyphoscoliosis and nystagmus in some patients18,20.
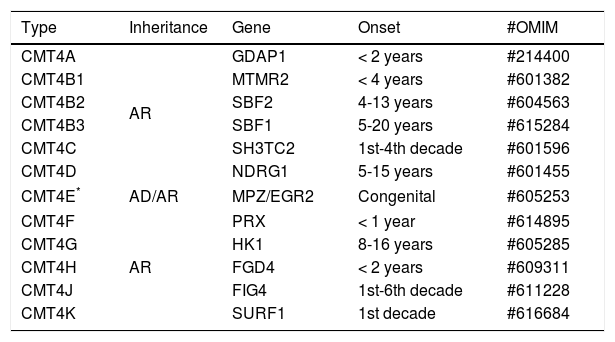
CHARCOT-MARIE-TOOTH DISEASE TYPE 4CMT4 represents a rare form of CMT. Its subtypes are described in a few families of specific ethnic groups in North Africa or in genetically isolated European populations21 (Table 3).
Classification of CMT type 4.
| Type | Inheritance | Gene | Onset | #OMIM |
|---|---|---|---|---|
| CMT4A | AR | GDAP1 | < 2 years | #214400 |
| CMT4B1 | MTMR2 | < 4 years | #601382 | |
| CMT4B2 | SBF2 | 4-13 years | #604563 | |
| CMT4B3 | SBF1 | 5-20 years | #615284 | |
| CMT4C | SH3TC2 | 1st-4th decade | #601596 | |
| CMT4D | NDRG1 | 5-15 years | #601455 | |
| CMT4E* | AD/AR | MPZ/EGR2 | Congenital | #605253 |
| CMT4F | AR | PRX | < 1 year | #614895 |
| CMT4G | HK1 | 8-16 years | #605285 | |
| CMT4H | FGD4 | < 2 years | #609311 | |
| CMT4J | FIG4 | 1st-6th decade | #611228 | |
| CMT4K | SURF1 | 1st decade | #616684 | |
AD: autosomal dominant, AR: autosomal recessive.
CMT4 is characterized by severe early-onset sensory-motor polyneuropathy, usually before two or three years of age. Clinical progression is rapid and distal limb deformity occurs markedly, with occurrence of talipes equinovarus, “clawed hands” and important deformities involving the spine. Depending on the subtype, electrophysiological and histopathological studies may demonstrate features of demyelination, in most of the cases, or primary axonal damage18,22.
The inheritance pattern is usually autosomal recessive. Mutation-related disease forms in the PMP22, MPZ and EGR2 genes are most commonly found in children with early hypotonia and respiratory failure (also being classified as Dejerine-Sottas disease), while mutations in GDAP1, MTMR2, SBF2, PRX, SH3TC2 or FGD4 are usually present in a later onset, with variable delay of motor development and deformity in feet, preceded by a neonatal period, mostly without intercurrences21,22.
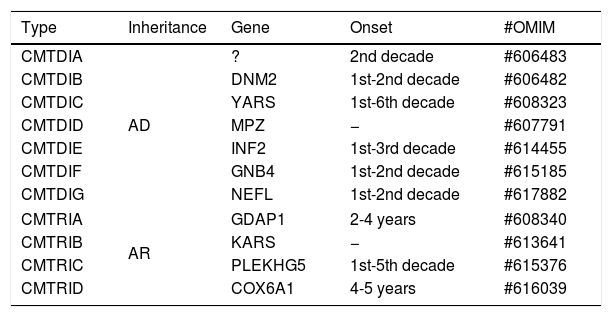
INTERMEDIATE FORMS OF CHARCOT-MARIE-TOOTH DISEASEThe studies related to the pathophysiological understanding of CMT allow us to divide it into two main groups: that of demyelinating neuropathies, which present with reduced nervous conduction velocity (NCV), and that of axonal neuropathies, with NCV close to normal (Table 4). However, there is a group of neuropathies that present NCV with intermediate values, overlapping the two other main groups.
Classification of intermediate forms of CMT.
| Type | Inheritance | Gene | Onset | #OMIM |
|---|---|---|---|---|
| CMTDIA | AD | ? | 2nd decade | #606483 |
| CMTDIB | DNM2 | 1st-2nd decade | #606482 | |
| CMTDIC | YARS | 1st-6th decade | #608323 | |
| CMTDID | MPZ | − | #607791 | |
| CMTDIE | INF2 | 1st-3rd decade | #614455 | |
| CMTDIF | GNB4 | 1st-2nd decade | #615185 | |
| CMTDIG | NEFL | 1st-2nd decade | #617882 | |
| CMTRIA | AR | GDAP1 | 2-4 years | #608340 |
| CMTRIB | KARS | − | #613641 | |
| CMTRIC | PLEKHG5 | 1st-5th decade | #615376 | |
| CMTRID | COX6A1 | 4-5 years | #616039 | |
AD: autosomal dominant, AR: autosomal recessive.
The cases of the intermediate forms of CMT can be recognized in families in which different individuals present with motor nerve conduction velocities (mNCV) varying from both type 1 and type 2 CMT, that is, above and below 38 m/s (usually between 25 and 45 m/s). It is important to emphasize that the term “intermediate” should not be used to describe a single measure of NCV, it is used to describe forms of disease that have characteristics in the pathology of both demyelinating and axonal damage13. The inheritance pattern in these cases is usually autosomal dominant but may also be autosomal recessive or X-linked23.
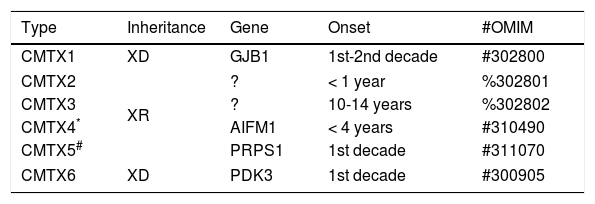
CHARCOT-MARIE-TOOTH DISEASE FORMS LINKED TO THE X CHROMOSOMECMT forms linked to the X chromosome (CMTX) represent about 7 to 10% of all cases of hereditary sensory-motor neuropathies (Table 5). Described initially in 1888, CMTX classically presents as hereditary neuropathies with phenotypes similar to CMT1, but which do not present direct transmission between men. It is important to remember that in these cases all the daughters of the parents bear the genetic defect, which is transmitted to half of their descendants24,25.
Classification of X-linked CMT.
| Type | Inheritance | Gene | Onset | #OMIM |
|---|---|---|---|---|
| CMTX1 | XD | GJB1 | 1st-2nd decade | #302800 |
| CMTX2 | XR | ? | < 1 year | %302801 |
| CMTX3 | ? | 10-14 years | %302802 | |
| CMTX4* | AIFM1 | < 4 years | #310490 | |
| CMTX5# | PRPS1 | 1st decade | #311070 | |
| CMTX6 | XD | PDK3 | 1st decade | #300905 |
XD: X-linked dominant, XR: X-linked recessive.
CMTX type 1 (CMTX1) is the major representative of this group and is caused by the mutation in the gene responsible for GJB1 protein (also known as connexin 32). This component is responsible for forming junctions between the different layers of the myelin. CMTX1 is considered a dominant X-linked condition, that is, female mutation carriers may also express neuropathy phenotype. However, in these cases, symptoms usually begin after the second decade of life and are usually milder than in males. In contrast, the other types of CMTX described have a pattern of X-linked recessive inheritance24,25.
DIAGNOSISThe approach for the diagnosis of CMT subtypes and other hereditary neuropathies depends on a line of clinical-laboratory reasoning that begins with the definition of the phenotype, identification of the inheritance pattern, electrophysiological study, and ends with molecular analysis. Nerve biopsy may be necessary in selected cases11.
The typical clinical phenotype, described in previous sessions, may be associated with most cases of CMT. Various clinical and physical examination findings may be useful in guiding molecular investigation, including age of onset, severity of disease, and presence of association with unusual symptoms.
The autosomal dominant inheritance pattern is the most commonly found and is seen in cases of CMT1 and in most cases of CMT2. It is important to remember that CMTX1 is transmitted in a dominant way linked to X chromosome, which is characterized by the absence of transmission between men, with women carrying the mutation in heterozygosis presenting a milder phenotype than the male individuals. Sporadic cases may occur and represent a diagnostic challenge, such as de novo mutations, which are particularly associated with CMT1A (by duplication of PMP22) and mutation of MFN2 (related to CMT2A2) but may also occur in other types of CMT11,26.
Family history, in some cases, may be less elucidative, since there is great variability of gene expression with many oligosymptomatic carriers. Summoning relatives of the patient referred to as “healthy” for performing electrophysiological test and a detailed physical examination may reveal important findings11.
ELECTROPHYSIOLOGICAL EVALUATIONThe electrophysiological test corresponds to an important step in the evaluation of individuals with suspected hereditary neuropathy and may be necessary for the planning of genetic tests. The study of nerve conduction (NCS) corresponds to the pillar of electrophysiological investigation in these cases. The main objective is to differentiate between demyelinating and axonal forms, or even to seek evidence for intermediate types27. The NCS can be easily performed in the relatives of the index case helping to define the pattern of inheritance.
For NCS at least three sensory nerves (sural, median and ulnar nerves) and three motor nerves (fibular, tibial and medial nerves) should be examined. Ideally, the evaluation should be supplemented with needle electromyography and somatosensory evoked potential27.
The forms CMT1 and CMT4 (autosomal dominant and recessive, respectively) present a very similar electrophysiological profile. The main feature in these forms is the marked reduction in mNCV below 38 m/s in the nerves of the upper limbs (usually using the ulnar nerve as a reference). Classically, mNCV values find their nadir up to five years of age. In CMT1, the mean value of mNCV is typically around 25 m/s, but with values ranging from 9 m/s to 41 m/s. In general, CMT4 usually presents with mNCV lower than CMT1. SNAP may be absent, especially in lower limbs, with CMAP usually quite reduced or absent14,28.
In CMT2 axonal forms, mNCV typically are above 38 m/s, contrasting with demyelinating variants. Correlating with axonal damage, electromyographic studies demonstrate increased motor unit potentials, fasciculation potentials, fibrillation potentials and positive acute waves. SNAP and CMAP are significantly reduced14,28.
The information available on the electrophysiological pattern in the DSD comes mostly from case reports, given the rarity of this condition and the difficulty of performing the complementary exam in these patients. The nerves of children with DSD are characterized by a high limit for stimulation, requiring the use of a higher and prolonged electrical current than usual for the stimulus. The mNCV in these cases is greatly diminished, usually with values lower than 10 m/s in the upper limbs. CMAPs usually present a significant reduction, and SNAPs are typically absent20.
The members of families bearing the intermediate forms of CMT present with mNCV that transit between the values of CMT1 and CMT2, usually between 25 and 45 m/s. The amplitude of CMAP is generally quite small13.
In cases of CMTX (represented mainly by CMTX1), the electrophysiological pattern may demonstrate characteristics of both demyelination and axonal damage. The mNCV in the upper limbs vary between 18 and 60 m/s, and the same individual may present different velocities on different nerves tested. SNAP and CMAP are also reduced24,27.
In addition, some features may help to differentiate hereditary neuropathies from acquired neuropathies, notably that temporal dispersion and conduction block are generally absent in inherited forms29.
NERVE BIOPSYPrior to the accessibility of the molecular test, the histopathology of the peripheral nerve was the most important tool in the diagnostic process of hereditary neuropathies. Currently, indications for peripheral nerve biopsy are restricted to select cases, in which diagnostic determination was not possible, despite a broad investigation of the mutations of the most commonly involved genes. Another possible scenario is one in which there is an important suspicion of sporadic hereditary neuropathy, but, however, causes of acquired neuropathies need to be excluded11,27.
The diagnostic value of nerve biopsy is influenced by factors such as the small amount of material usually extracted, the few options of nerves in the body suitable for sample collection, and the availability of specialized laboratories for tissue analysis27.
The nerve commonly chosen for the collection of material is the sural nerve, because besides being exclusively sensitive, it also has the necessary characteristics for a successful procedure: it is a superficial structure in which the cutaneous access is facilitated, usually it is affected by the pathological process and does not present technical difficulties to perform the pre-biopsy electrophysiological evaluation. Alternatively, the radial cutaneous nerve and the superficial fibular nerve can also be options27.
In CMT1 and CMT4, the demyelinating nature of the pathological process is confirmed by the occasional presence of large caliber axons devoid of myelin sheath and “onion bulb” formations in varying degrees, according to the specific type of CMT, more prominent in CMT1 forms. These formations reflect the concentric proliferation of the cytoplasm of Schwann cells around normally myelinated fibers27,30.
In axonal forms (classically CMT2 and CMTX) a reduction in the density of myelinated fibers is observed, mainly due to the compromise of the large caliber fibers. This reduction is more important in CMTX forms than in CMT2 forms. Irregular folds in the myelin sheath may be observed in some cases of CMT2. Non-myelinated fibers are relatively preserved and “onion bulb” formations may be seen27,30.
GENETIC TESTINGThe introduction of new generation sequencing (NGS) as a research tool in the field of hereditary neuropathies has allowed a progressive drop in the cost of whole-exome sequencing (WES). The trend of this evolution is to make many diagnostic algorithms by sequential testing of partially obsolete genes. However, it is important to remember that, despite the emergence of this new scenario, the cost of WES still remains quite high for use in the clinical routine31.
The success of molecular diagnostics shows advances in recent years due to the evolution of the new generation sequencing. Currently, approximately 80% of patients with CMT1 can receive an accurate molecular diagnosis, but a high percentage of cases of CMT2 (between 25% and 45%) remain unsolved32.
A sensible alternative is restricted sequencing, targeting only genes already known to be relevant to the disease. Such gene panels may be offered at a more competitive price but have the disadvantage of not identifying mutations in genes recently described31.
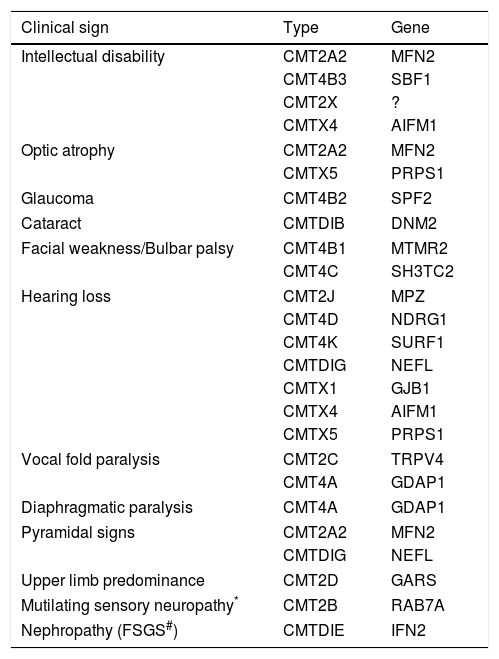
The traditional strategy of sequential gene testing is still a diagnostic option. The path to be followed in the algorithm depends on the mNCV and inheritance pattern and can be adapted if there are phenotypic characteristics that point to a specific molecular diagnosis, such as those described in Table 6. This approach has a reasonable success rate, with about 60% of patients reaching a genetic diagnosis, but it has a worse cost-benefit relationship as WES becomes popular33.
Uncommon clinical signals and symptoms in specific forms of CMT.
| Clinical sign | Type | Gene |
|---|---|---|
| Intellectual disability | CMT2A2 | MFN2 |
| CMT4B3 | SBF1 | |
| CMT2X | ? | |
| CMTX4 | AIFM1 | |
| Optic atrophy | CMT2A2 | MFN2 |
| CMTX5 | PRPS1 | |
| Glaucoma | CMT4B2 | SPF2 |
| Cataract | CMTDIB | DNM2 |
| Facial weakness/Bulbar palsy | CMT4B1 | MTMR2 |
| CMT4C | SH3TC2 | |
| Hearing loss | CMT2J | MPZ |
| CMT4D | NDRG1 | |
| CMT4K | SURF1 | |
| CMTDIG | NEFL | |
| CMTX1 | GJB1 | |
| CMTX4 | AIFM1 | |
| CMTX5 | PRPS1 | |
| Vocal fold paralysis | CMT2C | TRPV4 |
| CMT4A | GDAP1 | |
| Diaphragmatic paralysis | CMT4A | GDAP1 |
| Pyramidal signs | CMT2A2 | MFN2 |
| CMTDIG | NEFL | |
| Upper limb predominance | CMT2D | GARS |
| Mutilating sensory neuropathy* | CMT2B | RAB7A |
| Nephropathy (FSGS#) | CMTDIE | IFN2 |
There is currently no effective pharmacological therapy in CMT. Most of the disease management is related to rehabilitation therapy and surgical treatment of skeletal deformities. Patient follow-up should be multidisciplinary, including the assistance of neurologists, orthopedists, physiatrists, physiotherapists, and nurses.
Prescription of orthoses for lower limbs is recommended for patients with the classic CMT phenotype. Moderate physical exercises and stretching in order to avoid osteoskeletal complications are generally well tolerated. Patients with spine and limb deformities will commonly require corrective orthopedic surgeries34.
Fatigue can also be a complaint in CMT and is probably associated with different factors, including reduction of muscle strength and possible reduction in cardiorespiratory capacity. Obstructive sleep apnea syndrome is also common in CMT and its correction may have a positive response in improving fatigue in these patients35. Modafinil was a drug that showed benefit in this symptom in a small series of cases36.
THERAPEUTIC PERSPECTIVESIn CMT1A, the pharmacological strategies under study are aimed at modulating the overexpression of the PMP22 gene, since the decrease in the levels of the encoded protein has a positive effect on the CMT1A animal model. Previously, several substances have been studied for this purpose, such as ascorbic acid, but large clinical trials have failed to demonstrate significant beneficial effects on the outcome of the disease in humans.
PXT3003 (combination therapy of (RS) -baclofen, naltrexone hydrochloride and D-sorbitol) is the most advanced study compound for this line of therapy at the moment. This combination of drugs has demonstrated efficacy in promoting PMP22 down-regulation, with increased mNCV and improved motor dysfunction in animal models37. Currently, the PXT3003 clinical trial is in phase 3 (NCT02579759).
Despite the limited availability, antisense oligonucleotides (ASOs) have already proven their potential as a viable therapeutic option for use in the clinical practice of some diseases. In a natural line of evolution, recent studies of gene silencing with ASOs and small interfering RNAs that target the decrease in PMP22 expression have shown a response of significant electrophysiological and histopathological improvement in animal models38,39.
DISCLOSUREThe author declare no conflicts of interest whit this article.
4.