




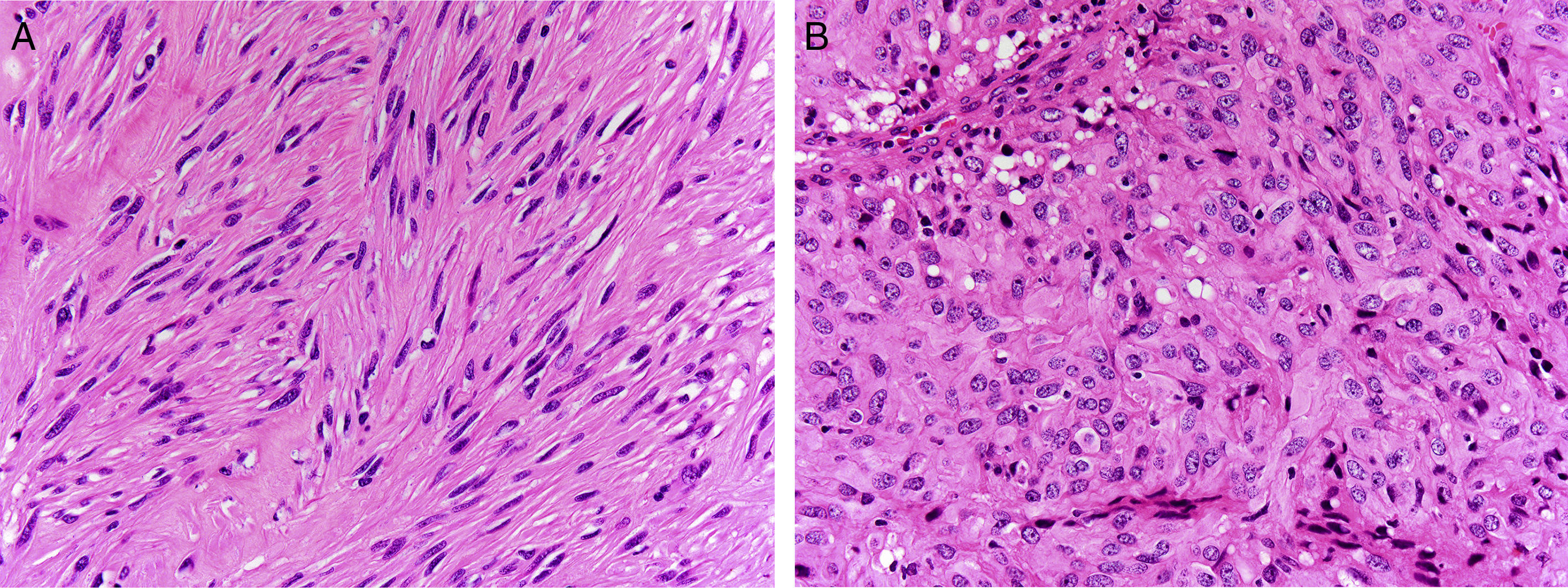
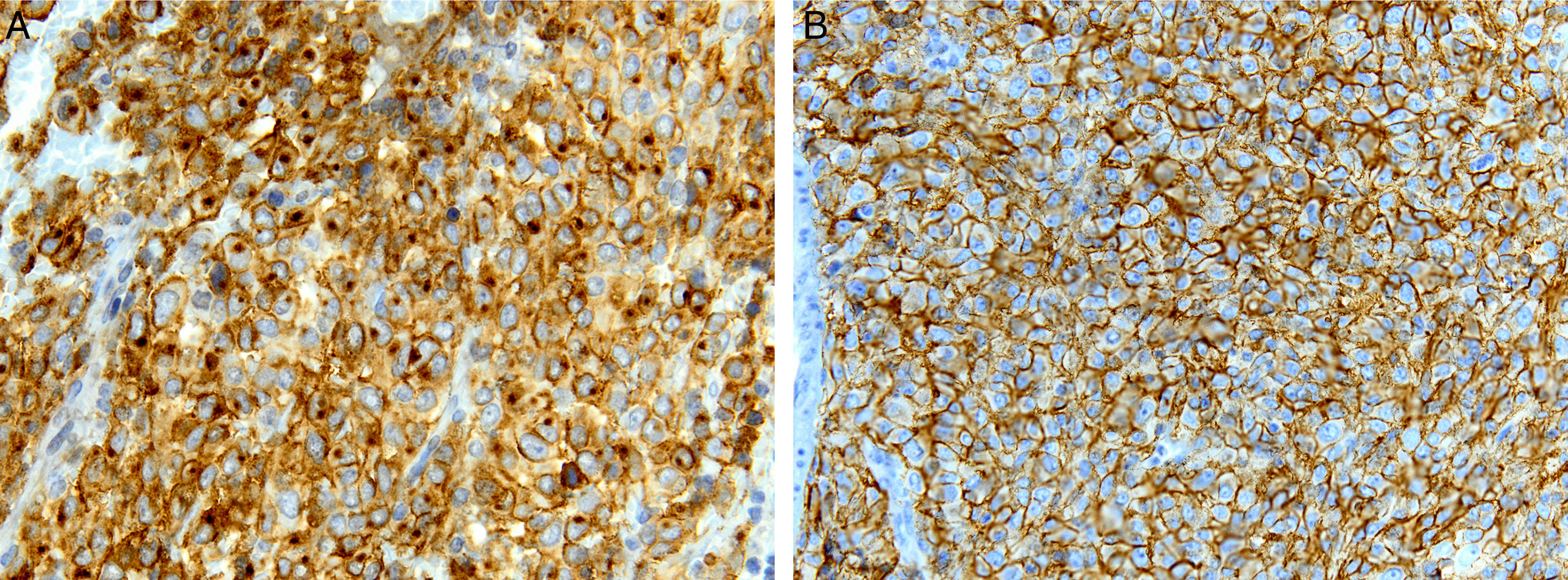


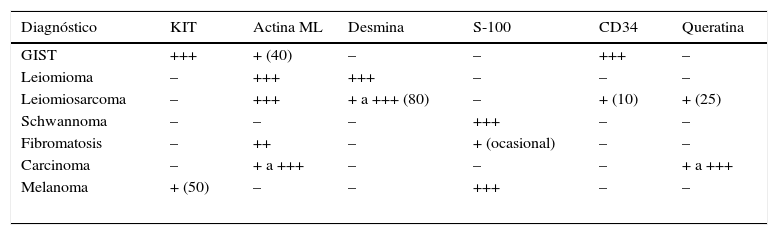
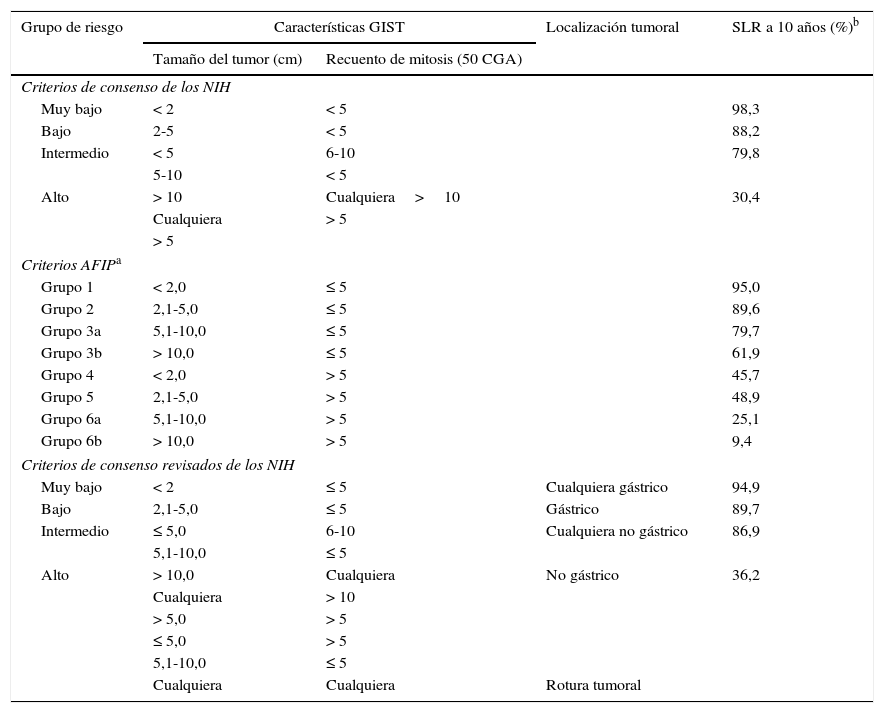
Los tumores del estroma gastrointestinal (GIST) son las neoplasias mesenquimales más frecuentes del tubo digestivo con una incidencia de 1,1 casos/100.000 habitantes/año. Un grupo de expertos de la Sociedad Española de Anatomía Patológica y la Sociedad Española de Oncología se reunieron para realizar una breve actualización de los GIST y consensuar los aspectos relacionados con su diagnóstico patológico y molecular. Los GIST generalmente son lesiones solitarias, bien circunscritas, de dimensiones variables (< 10 mm-35cm), que pueden presentar crecimiento parietal intra o extraluminal o mixto (en reloj de arena). Histológicamente, son neoplasias no encapsuladas, con crecimiento expansivo y celularidad fusiforme (70%), epitelioide (20%) o mixta (10%). La actividad mitótica generalmente es moderada o baja y debe evaluarse exclusivamente en los territorios con mayor celularidad o con mayor número de mitosis. La gran mayoría de los GIST presentan mutaciones activadoras, mutuamente excluyentes, en los genes que codifican para los receptores tirosina quinasa de tipo iii KIT y PDGFRA; con mucha menor frecuencia también se han descrito mutaciones en otros genes, tales como BRAF, NF1, y genes del complejo SDH. El método de detección de mutaciones de KIT y PDGFRA más ampliamente utilizado es la amplificación mediante reacción en cadena de la polimerasa de los exones de interés y la secuenciación directa (método Sanger) de estos productos de amplificación. Los informes moleculares siempre deben especificar el tipo de análisis llevado a cabo y la región o mutaciones que se han evaluado, así como indicar la sensibilidad del método de detección empleado.
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the digestive tract, with an incidence of 1.1 cases/100,000 inhabitants/year. A group of experts from the Spanish Societies of Pathology and Oncology met to discuss a brief update on GISTs and agree on aspects relating to the pathological and molecular diagnosis of these tumors. GISTs are generally solitary, well-circumscribed lesions of variable size (<10 mm-35cm) that may present with intra- or extra-luminal parietal growth or a mixed-type (hourglass) growth pattern. Histologically, they are unencapsulated neoplasms displaying expansive growth and spindle-shaped (70%), epithelioid (20%) or mixed cellularity (10%). Mitotic activity is generally moderate or low and should be evaluated only in areas with high cellularity or higher mitotic frequency. The great majority of GISTs harbour mutually-exclusive activating mutations in genes coding for the type iii receptor tyrosine kinases KIT and PDGFRA; less commonly, GISTs have also been reported to display mutations elsewhere, including BRAF and NF1 and SDH-complex genes. The method most widely used to detect KIT and PDGFRA mutations is amplification of the exons involved by polymerase chain reaction followed by direct sequencing (Sanger method) of these amplification products. Molecular analyses should always specify the type of analysis performed, the region or mutations evaluated and the sensitivity of the detection method employed.
Artículo

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora

