




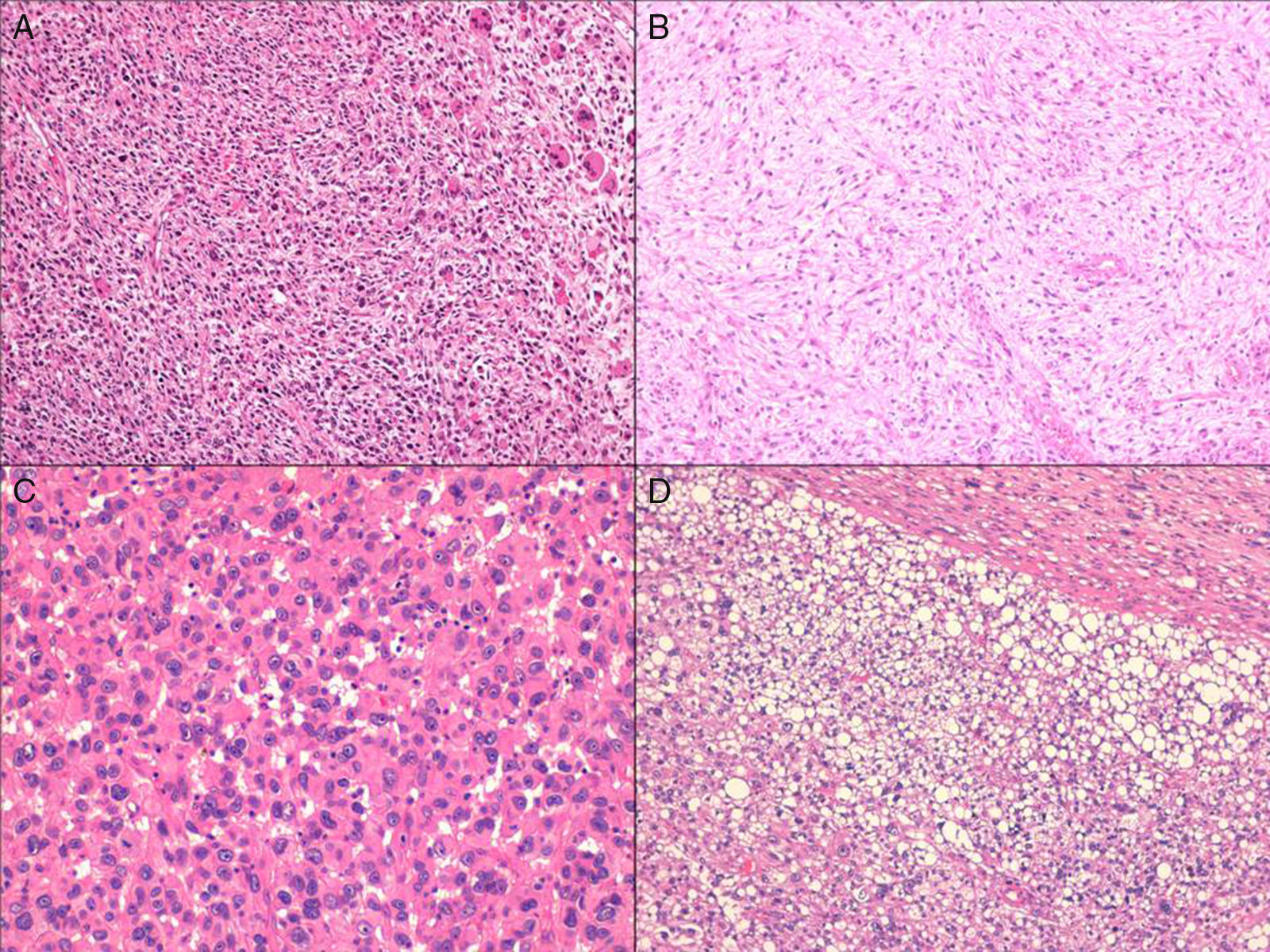

El liposarcoma pleomórfico es un sarcoma de alto grado que ocurre generalmente en la sexta-séptima décadas de la vida, afecta principalmente a partes blandas profundas de las extremidades inferiores y muestra una amplia variedad de patrones morfológicos, por lo que puede confundirse con otras lesiones tanto adipocíticas como no adipocíticas. La identificación definitiva de lipoblastos pleomórficos, que pueden ser muy escasos, es un requisito indispensable para el diagnóstico, por lo que es recomendable un muestreo amplio del tumor.
Pleomorphic liposarcoma is a high grade sarcoma occurring generally in the sixth to seventh decades of life and mainly affects the deep soft tissue of the lower extremities. As it can show a wide variety of morphologic patterns, it may be confused with other adipocytic and non adipocytic lesions. Definitive identification of pleomorphic lipoblasts is indispensable for diagnosis; however, as they can be very scarce, extensive sampling of the tumor is recommended.
Artículo

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora

