




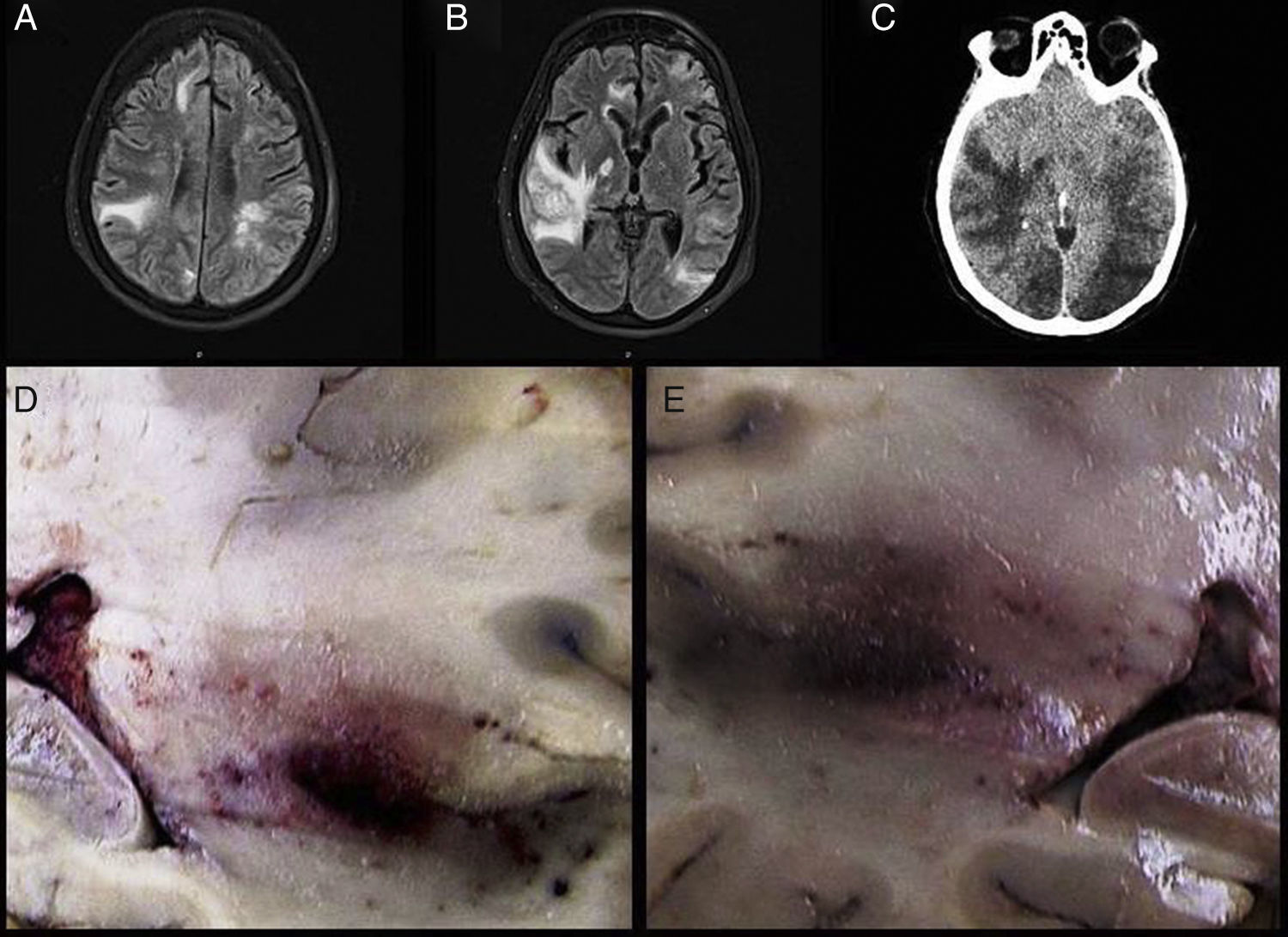
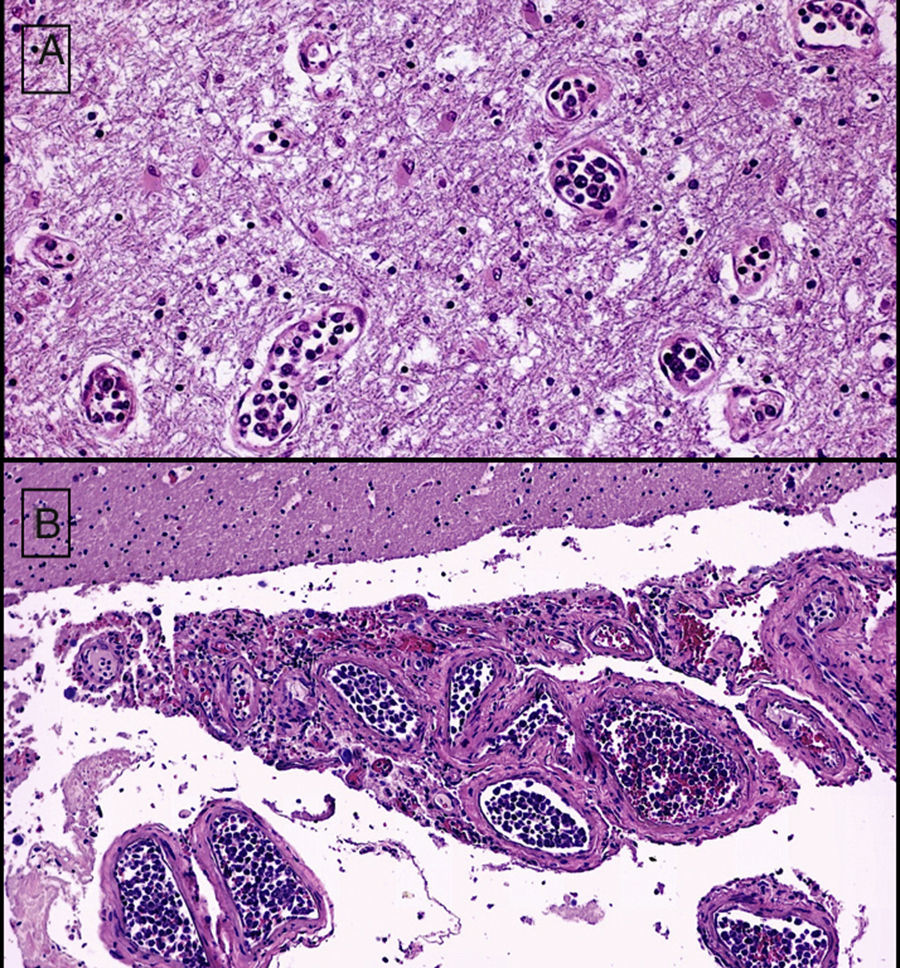
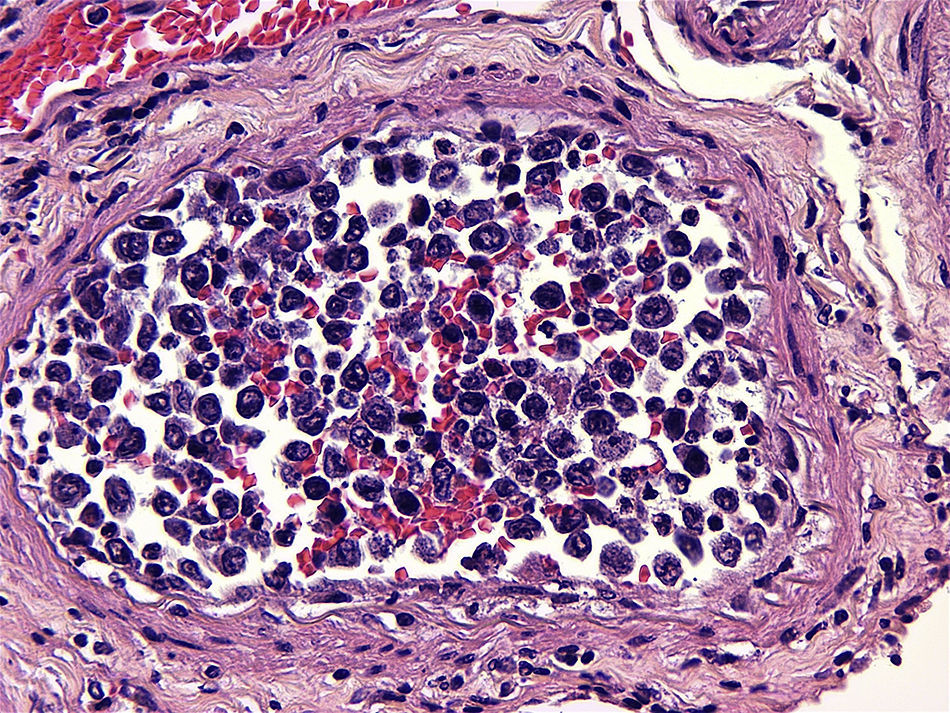
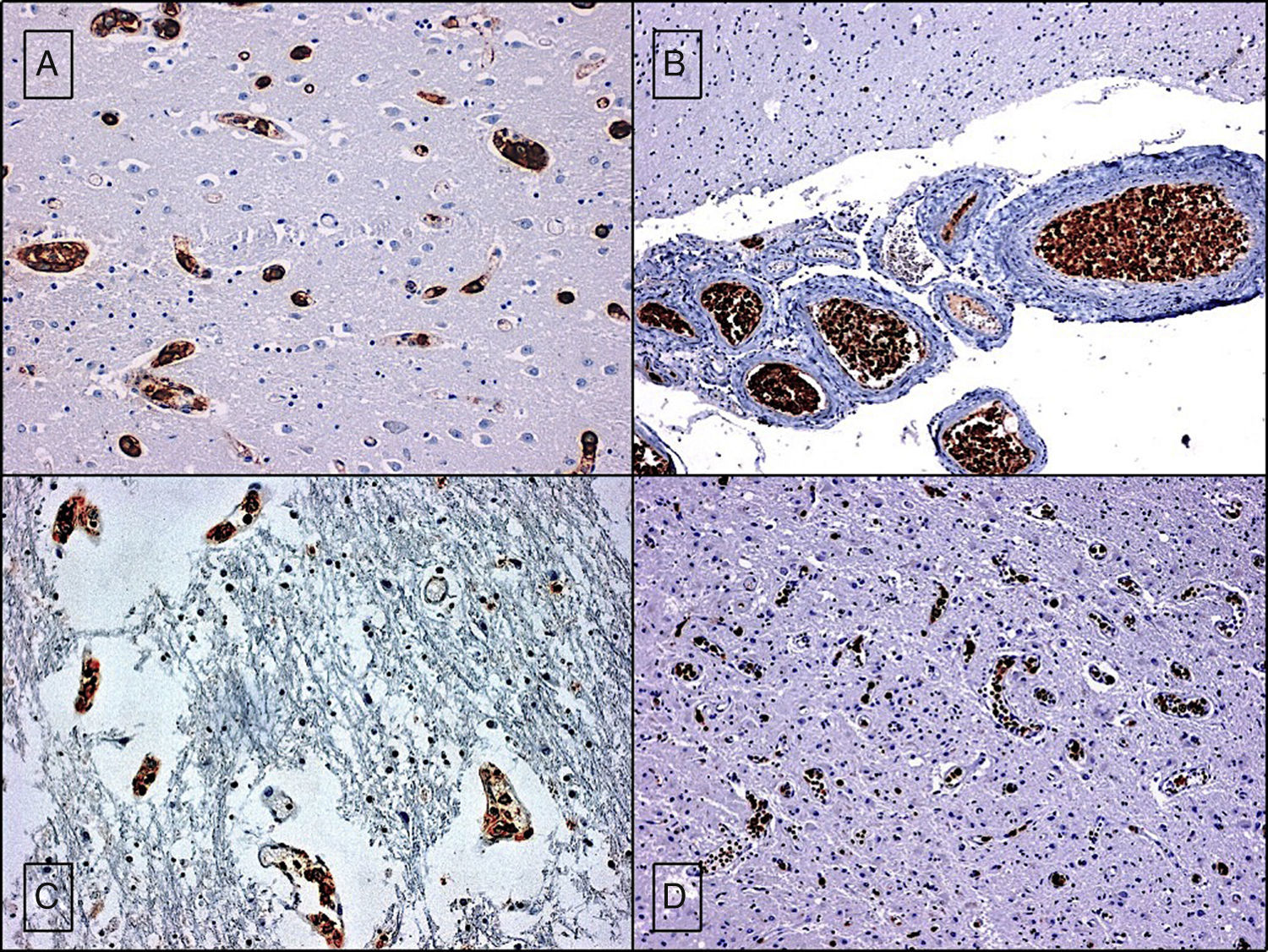
El linfoma intravascular es un tipo de linfoma extraganglionar, generalmente de células B, definido por una proliferación de linfocitos atípicos dentro de la luz de vasos de pequeño y mediano calibre. Desde su descripción en 1959 ha recibido diferentes denominaciones. En la actualidad, la Organización Mundial de la Salud se refiere a esta entidad como linfoma intravascular de células B grandes. Presentamos un caso caracterizado por deterioro neurológico de evolución rápida y progresiva consecutivo a eventos isquémicos multifocales y recurrentes de etiología inicialmente indeterminada. En el estudio post mortem limitado a la cavidad craneal se detectó una proliferación celular atípica en la luz de vasos de pequeño y mediano calibre. Con técnicas inmunohistoquímicas se confirmó el origen linfoide de las células neoplásicas intravasculares y se estableció el diagnóstico de linfoma intravascular de células B grandes.
Intravascular lymphoma is a type of extranodal lymphoma, usually composed of B-cells, resulting from a proliferation of atypical lymphocytes within the lumina of small to medium sized vessels. Since its initial description in 1959, it has had many names but currently the World Health Organization refers to this entity as intravascular large B-cell lymphoma. We present a case which presented with rapid progressive neurological deterioration and consecutive progressive multifocal and recurrent ischemic events of unknown origin. The postmortem study of the cranial cavity revealed atypical cell proliferation within the lumina of small to medium sized vessels. The lymphoid origin of intravascular tumor cells was confirmed by immunohistochemistry, establishing a diagnosis of intravascular large B-cell lymphoma.
Artículo

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora

