




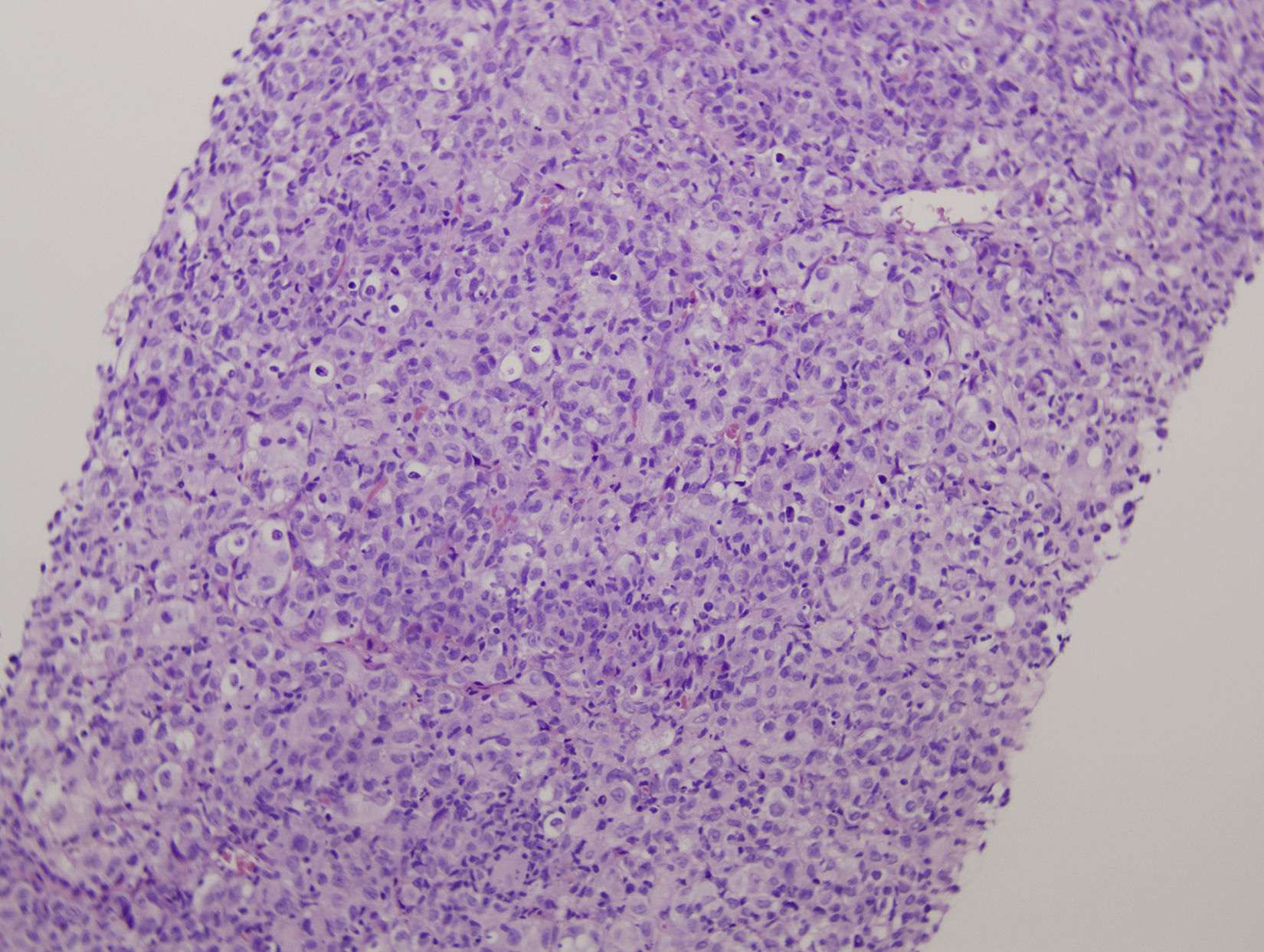
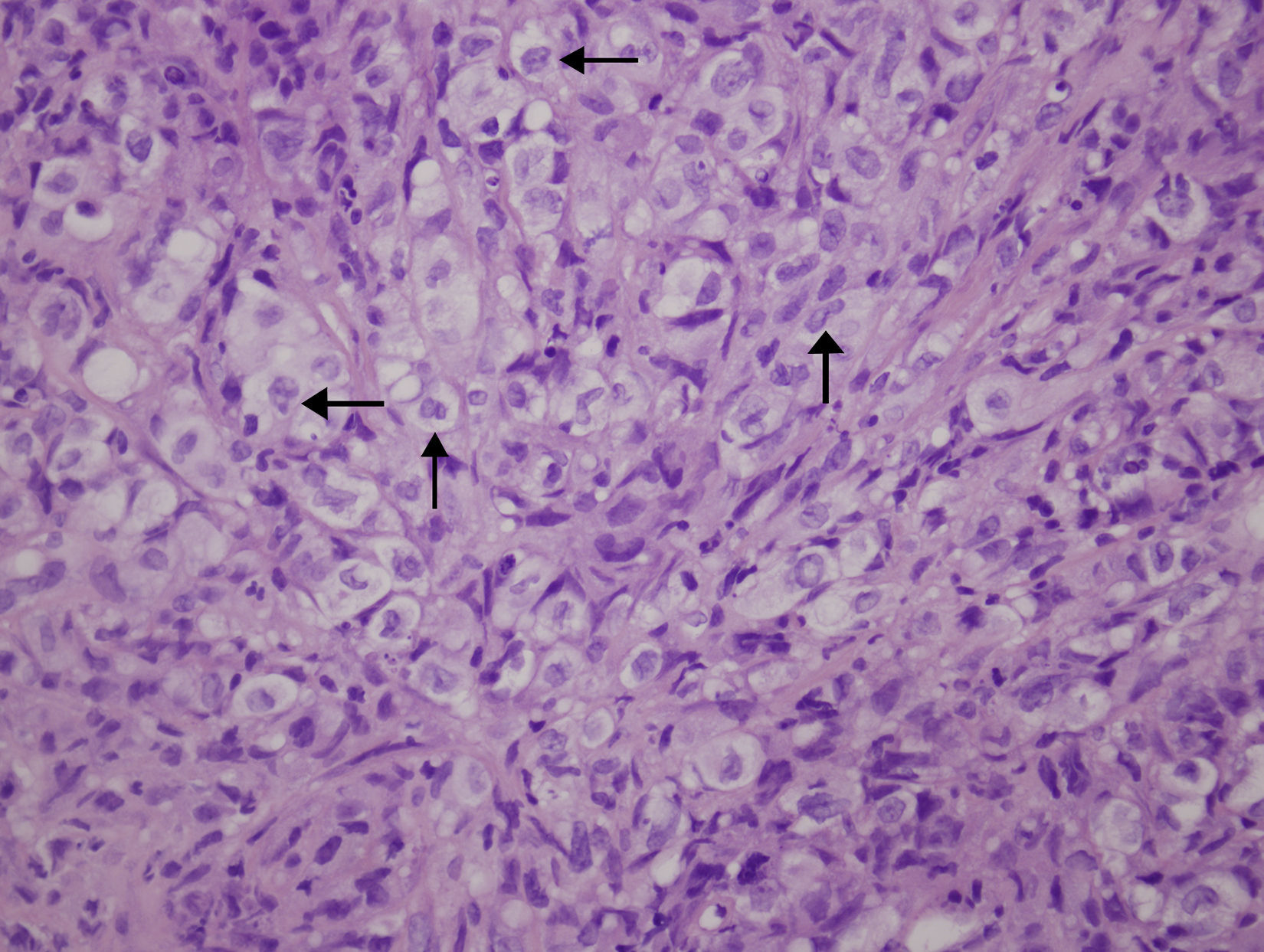
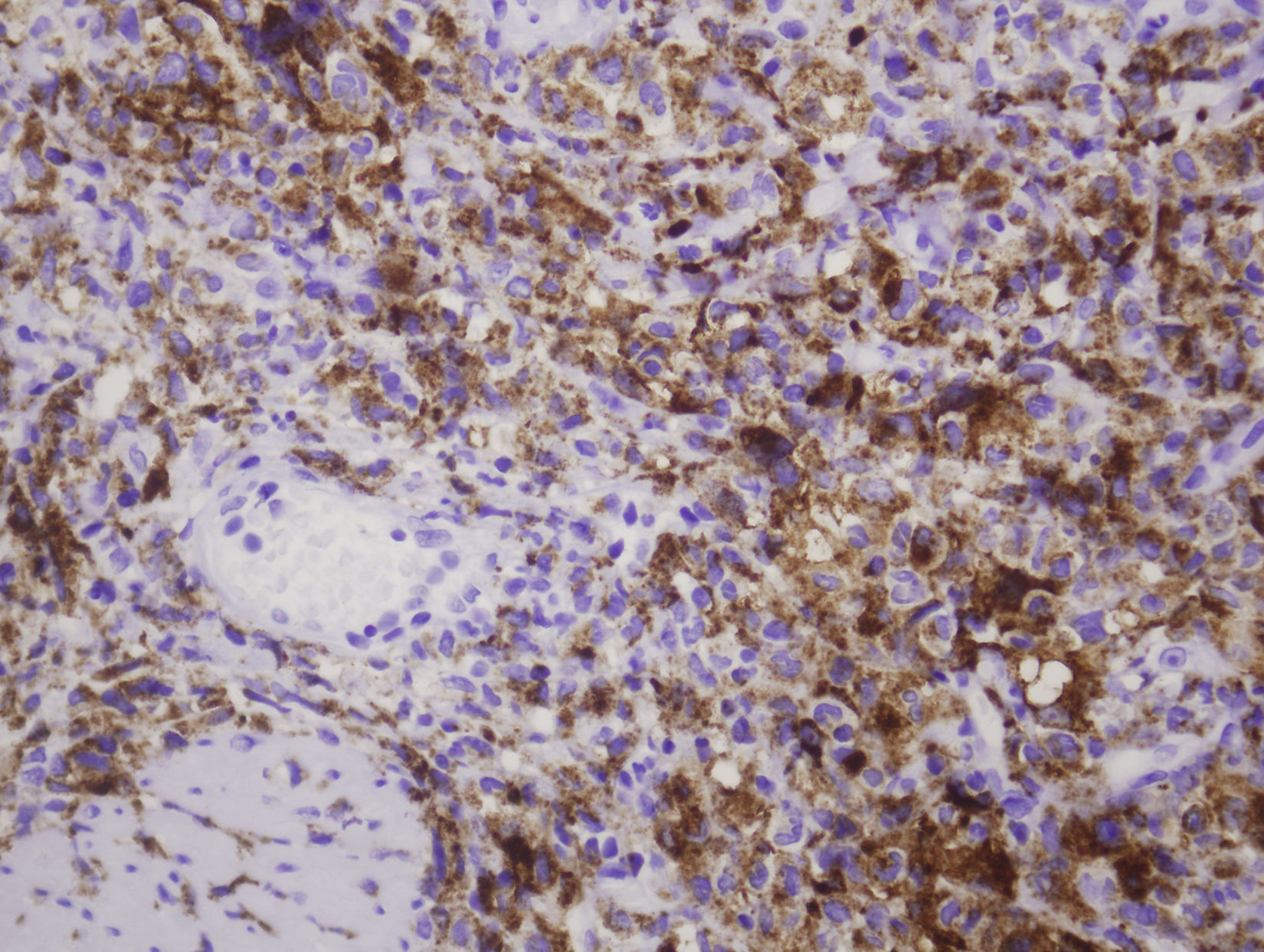
We report an unusual case of histiocytic sarcoma with bladder involvement. An 80 year-old man with a previous history of diffuse large B-cell malignant lymphoma presented with hematuria and back pain. Serial urine cytologies revealed no urothelial malignant cells, but cystoscopy showed a large intravesical mass. The patient underwent transurethral resection (TUR) of the tumor. The bladder TUR specimen showed a widely infiltrating epithelioid neoplasm, with intense immunohistochemical positivity for CD45 and histiocytic markers (CD68, lysozime and fascin). Histopathological diagnosis was histiocytic sarcoma. As the patient's condition was progressively deteriorating, only palliative care was indicated and he died one month after TUR. Although histiocytic sarcoma can often be widespread at the time of diagnosis, to our knowledge, this is the first report of a case presenting with urinary symptoms. Histiocytic sarcoma can mimic many other malignant lesions, and only immunohistochemistry can define the tumor cells, allowing correct therapy. We discuss the differential diagnosis and possible associations.
Presentamos un caso infrecuente de sarcoma histiocítico con afectación vesical. Varón de 80 años con historia previa de linfoma maligno difuso de células B grandes que presenta hematuria y dolor de espalda. Citologías seriadas de orina no mostraron células uroteliales malignas, pero una citoscopia reveló una gran masa intravesical. Se practicó una resección transuretral del tumor. La resección transuretral de la vejiga mostró una neoplasia tipo epitelioide con fuerte expresión de CD45 y de marcadores histiocíticos tales como CD68 lisozima y fascina con diagnóstico de sarcoma histiocítico. Tras el estudio, el paciente se deterioró progresivamente indicándose solo cuidados paliativos, con exitus al mes de la cirugía. Aunque el sarcoma histiocítico suele estar en estadio avanzado en el momento del diagnóstico, creemos que este caso es el primero en comenzar con síntomas urinarios. El sarcoma histiocítico puede simular diversas lesiones neoplásicas, siendo la inmunohistoquímica necesaria para un diagnóstico correcto. Discutimos diagnósticos diferenciales y posibles asociaciones.
Artículo

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora

