



The prevalence of bone dysplasias is estimated to be one case per 1000 inhabitants, which suggests that, at some point in the career of an orthopedic surgeon, he will face with one of these patients. The aim of this paper is to review the general aspects of bone dysplasias and focus on those, which due to their frequency and importance, we consider most relevant (achondroplasia, multiple epiphyseal dysplasia, spondyloepiphyseal dysplasia, osteogenesis imperfecta), reviewing their fundamental features and the latest therapeutic advances. There is no cure for these diseases, so early diagnosis and appropriate therapeutic management become the key to improving quality of life of these patients.
La prevalencia de las displasias óseas se calcula en un caso por cada 1.000 habitantes, lo que evidencia que, en algún momento de la trayectoria profesional de un especialista en cirugía ortopédica, se encontrará ante un paciente afecto por ellas. El objetivo de este trabajo es revisar los aspectos generales de las displasias óseas y centrarnos en las que, por su frecuencia e importancia, hemos considerado más destacadas (acondroplasia, displasia epifisaria múltiple, displasia espondiloepifisaria y osteogénesis imperfecta), revisando sus características fundamentales y los últimos avances terapéuticos. No existe tratamiento curativo para estas enfermedades, por lo que el diagnóstico precoz y el manejo terapéutico adecuado se convierten en la clave para mejorar la calidad de vida de estos pacientes.
The terms bone dysplasias, skeletal dysplasias and osteochondrodysplasias refer to a heterogeneous group of skeletal disorders which present the common feature of generalized alteration of bony and cartilaginous tissue. They are one of the most common causes of severe growth delay.
It is important to clarify certain concepts which tend to be used indistinctly:
- •
Dysplasia: processes involving generalized skeletal defects caused by intrinsic alterations.
- •
Dysostosis: processes limited to a specific bone or bone segment.
- •
Dystrophy: defects caused by an extrinsic process.
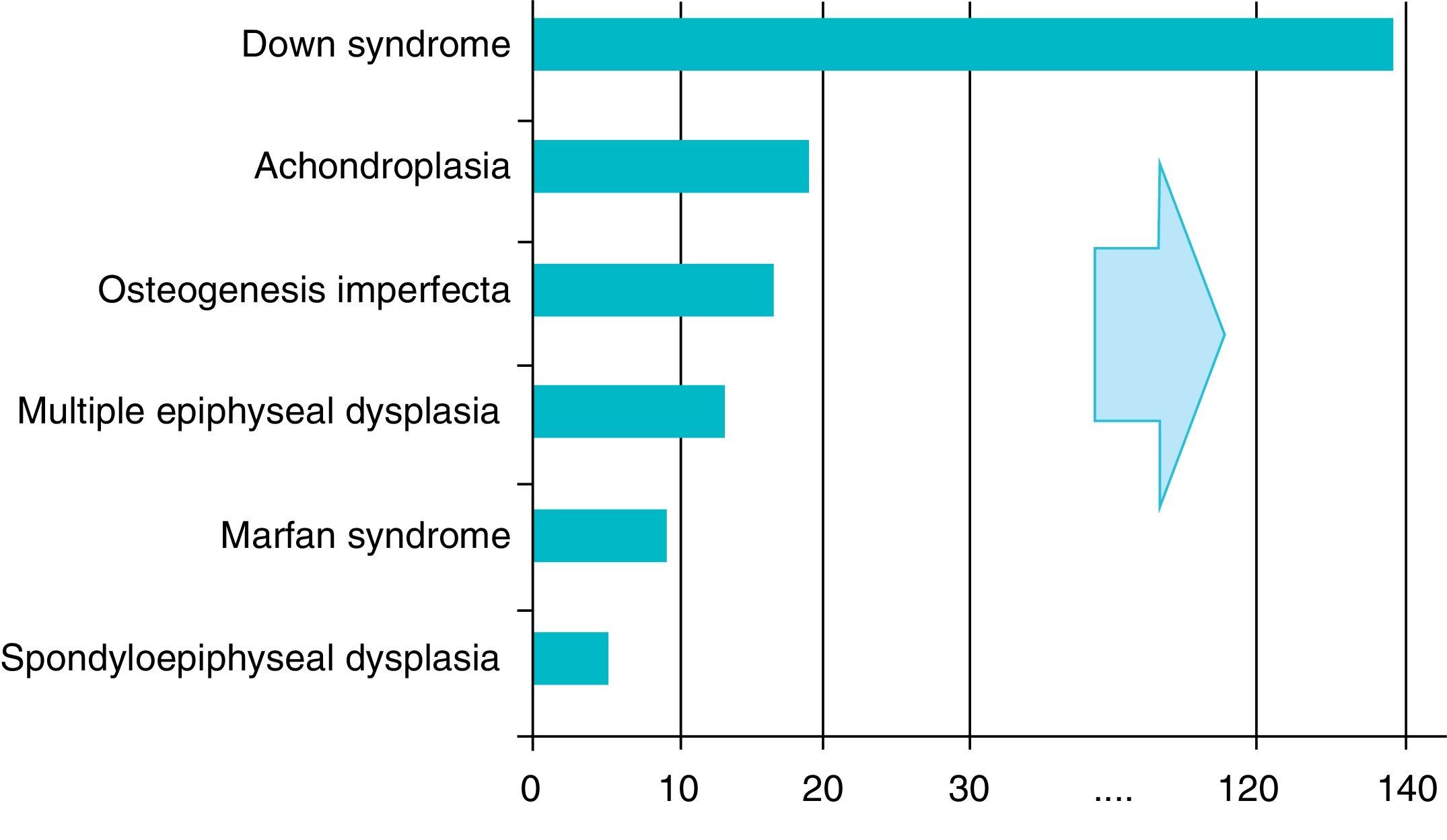
There are over 200 skeletal dysplasias, some of them with a very low prevalence. The overall frequency of these diseases is difficult to establish, but if we include latent or hidden forms which are not diagnosed, the prevalence would reach 1 case per 1000 inhabitants, with achondroplasia being the most common (Fig. 1).

The pathogenesis of these disorders can be attributed to a deficiency of normal tissue components, to an excess of mutant elements, to the accumulation of intermediary metabolites and to irregularities in the mechanisms which regulate the development of bony and cartilaginous tissue.1 The etiopathogenesis of these diseases is often linked to mutations in the genes encoding collagen proteins. Mutations in the COL2A1 gene have been described in chondrodysplasias with ocular involvement, such as spondyloepiphyseal congenital dysplasia and Kniest syndrome, mutations in the COL10A1 gene in Schmid metaphyseal dysplasia and mutations in the COL9A2 gene in multiple epiphyseal dysplasia (MED). Another frequently affected protein in these entities is fibroblast growth factor receptor 3 (FGFR3), which presents mutations in achondroplasia, hypochondroplasia and thanatropic dysplasia. Most bone dysplasias follow a pattern of autosomal dominant inheritance. However, many of them arise from de novo mutations, with advanced paternal age being a significant risk factor.
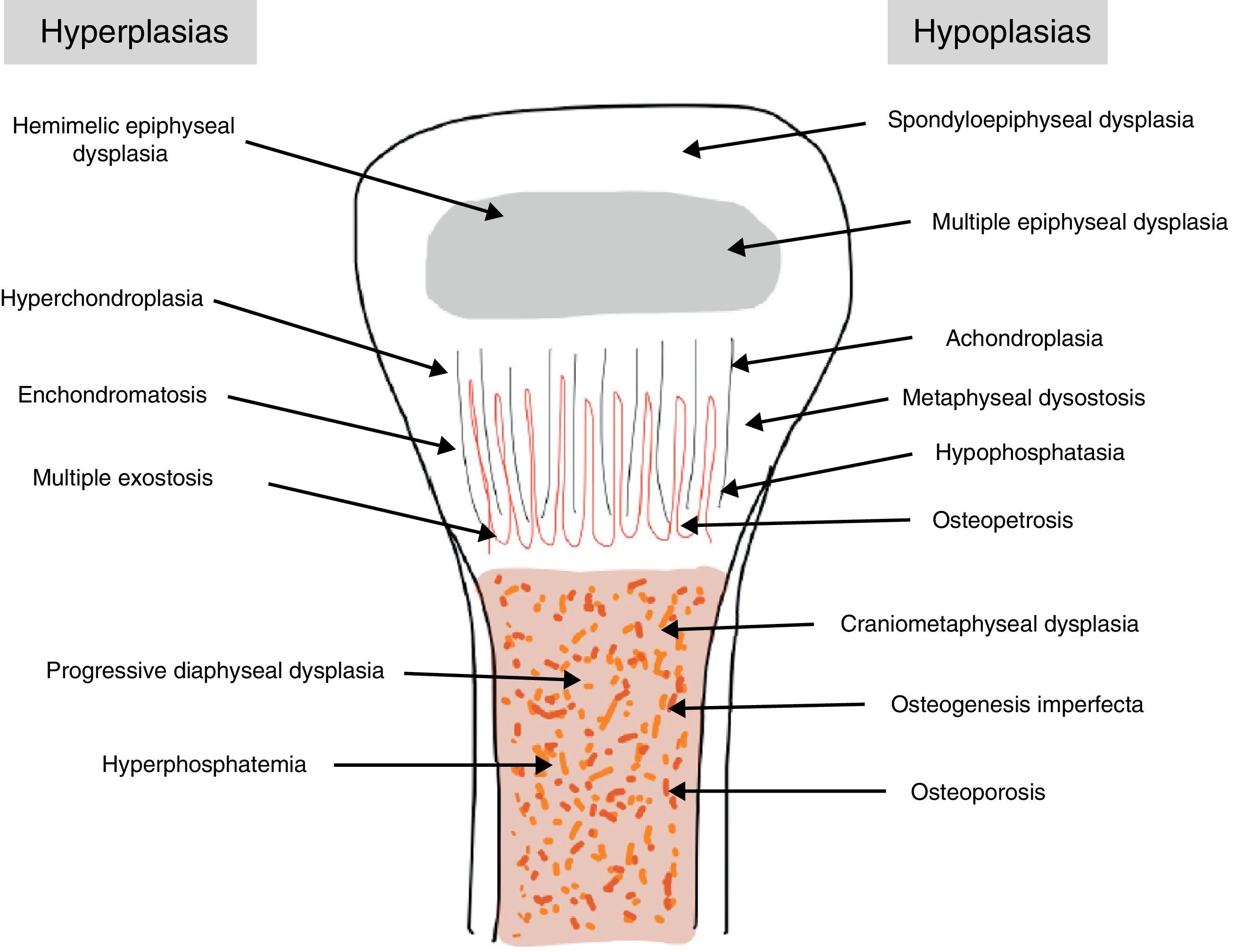
The traditional classification is that established by Rubin,2 which differentiates between hypo- and hyperplasias according to the affected bone area (Fig. 2). However, it is currently more common to group them according to their molecular origin and genetic disorders identified.3 One example is the classification established by Nelson4:
- •
Involvement of the cartilage matrix proteins: spondyloepiphyseal dysplasias, MED.
- •
Involvement of transmembrane receptors: achondroplasia group.
- •
Altered ion transport: diastrophic dysplasia, achondrogenesis.
- •
Involvement of transcription factors: campomelic dysplasia, nail-patella syndrome.
- •
Defects in bone resorption: osteopetrosis.
- •
Unknown defects: asphyxiating thoracic dystrophy or Jeune syndrome, Caffey disease or infantile cortical hyperostosis.

In daily clinical practice it is more practical to classify them by skeletal area affected: (a) axial skeleton (spondyloepiphyseal dysplasia [SED] and metatropic dysplasia or Kniest syndrome), (b) epiphysis (MED, hemimelic epiphyseal dysplasia) and (c) metaphysis (achondroplasia, thanatophoric chondrodysplasia).2,5
The main characteristics of the most prevalent1–5 and significant bone dysplasias in our environment are summarized below.
AchondroplasiaOverviewThis is the most common bone dysplasia, with an estimated incidence of 1/30,000 live newborns,2,5 and the best-known type of dwarfism.1–5 The term employed to describe the disease is not entirely appropriate, as it is a failure of endochondral ossification, rather than chondrogenesis.2
EtiologyAlthough it follows an autosomal dominant pattern, it appears de novo in up to 80% of cases4 due to a point mutation in codon 380 of the gene encoding FGFR3 located on the short arm of chromosome 4.1–5 FGFR3 seems to limit endochondral ossification and its mutation increases this inhibition,2 with intramembranous and periosteal ossification remaining unaltered.
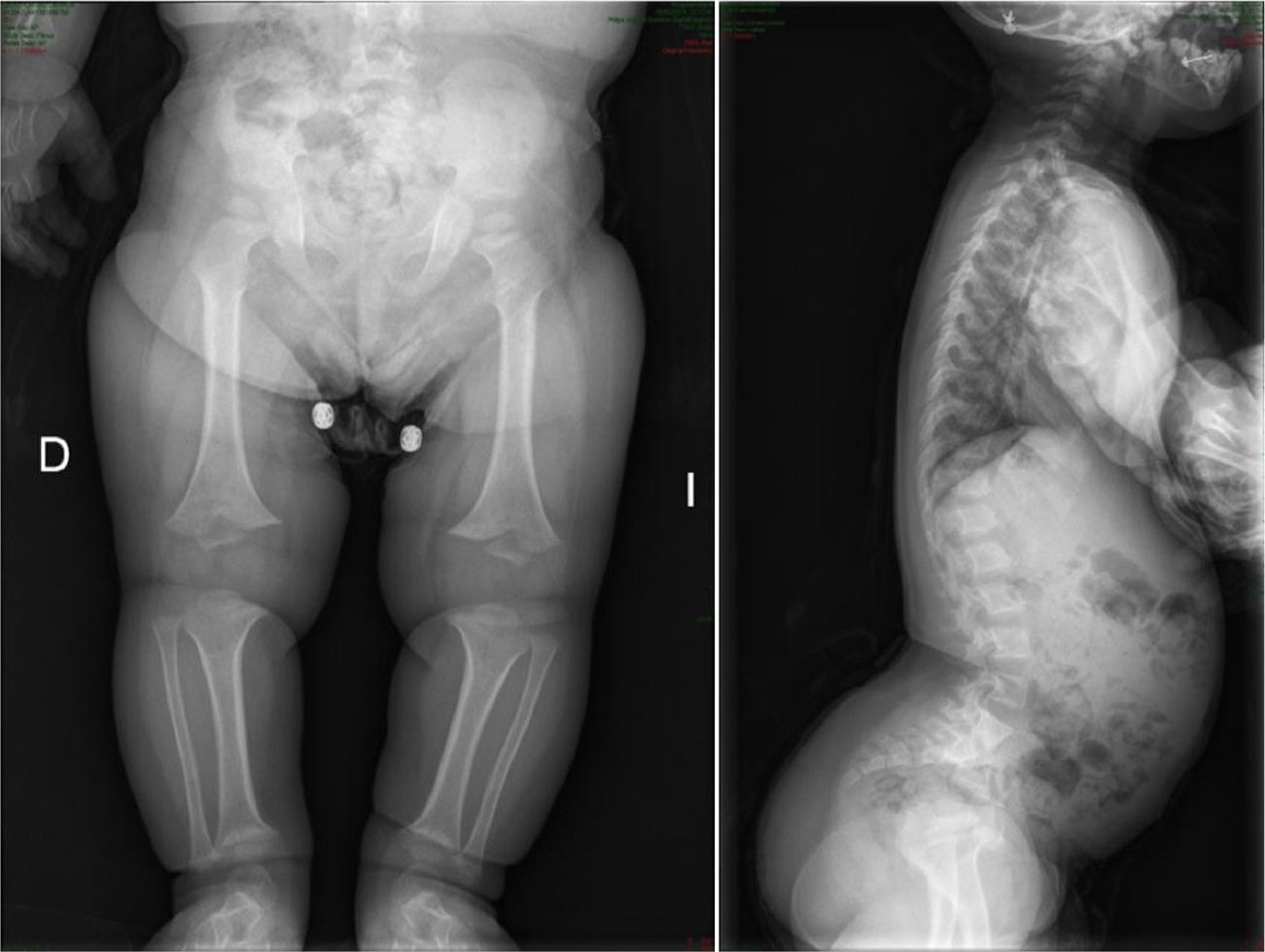
CharacteristicsIt is usually diagnosed at birth or prenatally,3 through observations of disproportionately short stature with characteristic craniofacial dysplasia (macrocephaly, frontal and biparietal prominence, midface hypoplasia, flattening of the occiput, depressed nasal bridge and prognathism1–5). It is important to distinguish it from other forms of dwarfism in the neonatal period.1 In this case, it is of rhizomelic type, where the proximal limb segments are relatively short in comparison with the trunk.1–5 Other characteristics which are present at birth include redundant skin folds with appearance of excessive muscle tissue, extreme lassitude of all joints except the elbow, which has a certain limitation for extension, short hands with “trident” deformity, called thus because there is a clear separation between the middle and ring fingers, and “sea star” deformity called thus because the 3 middle fingers have the same length. During lactation there is usually a delay in the acquisition of motor skills due to hypotonia, joint hyperlassitude and mechanical difficulty to balance an oversized head with a normal trunk and short limbs. However, this motor delay can also be the first manifestation of a cervical myelopathy resulting from stenosis of the foramen magnum, an entity which should always be ruled out in these patients.1,2,4 Subjects may present thoracolumbar kyphosis, which evolves into lumbar hyperlordosis as the child begins to walk.1–5 Hydrocephalus may appear, usually not requiring treatment, as well as repeated otitis media, which can lead to deafness, and dental crowding. The IQ of these patients is usually normal. Angular deformities appear in both lower limbs during childhood and adolescence: genu varus, internal tibial torsion and varus deviation of the ankle, due to tibial malformation and increased fibular length with respect to the tibia.1–5 Coxalgia and neurological symptoms in the lower limbs may appear in adulthood as a possible manifestation of spinal canal stenosis, which should not to be attributed to early osteoarthritis, which is infrequent.1
The radiological findings present at birth, along with the phenotype, are diagnostic; it is essential to request radiographic studies of the skull, lumbar spine and pelvis1 (Fig. 3). The cranial bones are large, whilst the skull base and facial bones are hypoplastic. Occipitalization of the atlas is frequently carried out in the cervical spine.1 The vertebral pedicles are short and the interpedicular distance is inverted.1,2,4 The ilia are short and rounded, with an appearance of “elephant ears”, the sacrum is horizontalized and the acetabular roofs are flat. Wedging of T12-L1 is not uncommon, eventually producing cauda equina syndrome.1 All tubular bones are short, except for the fibula which is disproportionately long compared to the tibia. The growth plate of the distal femur presents a characteristic, inverted “V” shape.1 The radial head is deformed and tilted toward the posterior part, degenerating into subluxation and/or posterior dislocation thereof in late childhood.

Early diagnosis, continuous neurological assessment (for foramen magnum stenosis during early childhood and lumbar in adulthood) and providing the family with information about the disease are very important.
The use of growth hormone has not shown great benefits,2,3 as the disease is not due to a deficit thereof,2 and positive results have only been obtained in patients with a lower growth rate.2 Recent studies have shown a decrease in cellular expression of parathyroid hormone receptor and an increased tendency of chondrocytes toward apoptosis, a fact that would explain the lack of response to growth hormone therapy.2,6
Orthopedic treatment is focused on 3 aspects: bone elongation, correction of axial deviations and treatment of spinal stenosis and thoracolumbar kyphosis. Bone elongation is the most controversial and, although it has been conducted frequently in recent years, at present there are doubts regarding the real benefits it provides. In clinical practice, this process only offers benefits if a final height of 150cm is achieved, implying elongations of 25–30cm in the lower limbs, as well as the need to also lengthen the humerus in order to facilitate personal care. The total period required may exceed 2 years and it entails a high risk of complications. Several authors have not found statistically significant differences in terms of improving quality of life in elongated patients compared to patients suffering achondroplasia who did not undergo this procedure.7–9 Seung-Ju et al.10 used 3 questionnaires and found that the scores obtained in the mental, physical and functional components were similar for both groups of patients and that these scores were inversely proportional to the number of complications arising during the elongation process. These authors concluded that bone elongation could be a reasonable treatment option provided that patients understand the benefits and risks of the process and that it is conducted as a multidisciplinary process paying special attention to any complications which arise so they can be acted on at an early stage. The Little People of America Association came to the same conclusions.11
As for the treatment of spinal deformities, kyphosis is the most common, mainly at the D12-L1 level, and stenosis is the most severe.1–5 In most cases, kyphosis improves when the child begins to walk, but in 10–15% of cases it remains and even increases. Treatment will depend on the age at which the deformity is detected. During childhood the main objective is to prevent the occurrence of the deformity through certain postural rules, such as avoiding sitting in backless chairs and maintaining a straight back in daily life. Once it is established, a brace is indicated if the kyphosis is accompanied by a progressive and significant vertebral wedging and/or if the Cobb angle is not reduced below 30° in hyperextension radiographs. In cases in which a normal brace is insufficient, a plaster brace may be applied in hyperextension. Surgical treatment is indicated for cases in which kyphosis persists despite conservative treatment. The diagnosis of spinal stenosis, normally obtained during the third decade of life by the presence of neurogenic claudication, represents an absolute and immediate indication for surgical decompression. This is carried out from several levels above the stenosed area to S2, with laminectomies and even foraminotomies being necessary in those cases in which the roots are also affected.2 Once this complication has been established, the results are unpredictable.1
Several lines of research are currently attempting to reduce the hyperactivity of FGFR3,12 including: selective chemical inhibition of tyrosine kinase activity in FGFR3,13 antibodies which block activation of FGFR3 based on trastuzumab,14,15 (monoclonal anti HER2/neu antibody) and the use of C-type natriuretic peptide as an antagonist of the signal triggered by FGFR3.16–18 Although this disease is associated to a high rate of perinatal mortality, those patients who survive have a similar life expectancy to that of the general population4 and their overall health assessment index is not significantly lower.2 The mean final height reached is 131cm among males and 124cm among females.1
Multiple epiphyseal dysplasiaOverviewMED is another example of the most common skeletal dysplasias caused by a disruption in endochondral ossification of the epiphysis, especially at the level of the proximal femoral epiphysis.3 It has a prevalence of 10–12 cases per million inhabitants.4 Conventionally, 2 variants of the disease were identified, the severe or Fairbank type form and a milder of Ribbing variant. At present, based on current knowledge of the genetic bases of this bone dysplasia, it is considered as a single disease, without variants, but with a highly variable expression.2
EtiologyIt is an autosomal dominant disorder, although the literature contains reports of recessive cases of this disease caused by a mutation in the gene encoding the oligomeric cartilage matrix protein located on chromosome 19. There are also mutations in the genes encoding type IX collagen (COL9A2) and matrilin-3.1,2,4 The end result of these mutations is an anomaly in the matrix of hyaline, articular and physeal cartilage1 resulting in the typical deformities of the disease.
CharacteristicsMED is not usually diagnosed until early childhood through consultation for one of the following reasons: joint pain in the lower limbs (mainly the hips), decreased range of mobility, gait alterations and angular deformities of the knees.2 Load bearing joints (hips, knees and ankles) are the most commonly affected. The hands of these patients are small and with impaired mobility, and fatigue of the hands caused by writing is a common trait.1 The growth rate is decreased, with a final result of short stature, although without reaching dwarfism.1–3 The facies and spine are normal. There are no visceral alterations and the level of intelligence is not affected. A prenatal diagnosis is possible in familial cases associated to cartilage matrix oligomeric protein.
Conventional radiology is essential for the diagnosis of the disease, and studies of the pelvis, spine, knees, ankles and wrists are also necessary.1Radiological findings may include delayed appearance of any ossification nucleus, although the changes will be most evident in the proximal and distal epiphyses of the femurs, and proximal of the humerus and tibias.2 The severity and extent of the disease are highly variable among families, which influences the age of onset of osteoarthritis. Radiological changes in the hips are reminiscent of those observed in Perthes disease, the main differential diagnosis of this bone dysplasia. In fact, MED should be ruled out in any patient suspected of suffering bilateral Perthes.1–3 The features that distinguish this entity from Perthes disease include symmetrical and fairly synchronous radiological changes in the hips, preserved femoral metaphyses and articular cartilage, more severe involvement of the acetabulum and absence of femoral head subluxation.2 SED is the other bone dysplasia which should be distinguished from MED, with involvement or not of the spine being the key point to establish this distinction.1 Osteoarthritis is the main problem of these patients and usually occurs between the second and third decades of life, depending on the severity of the disease, as mentioned previously.
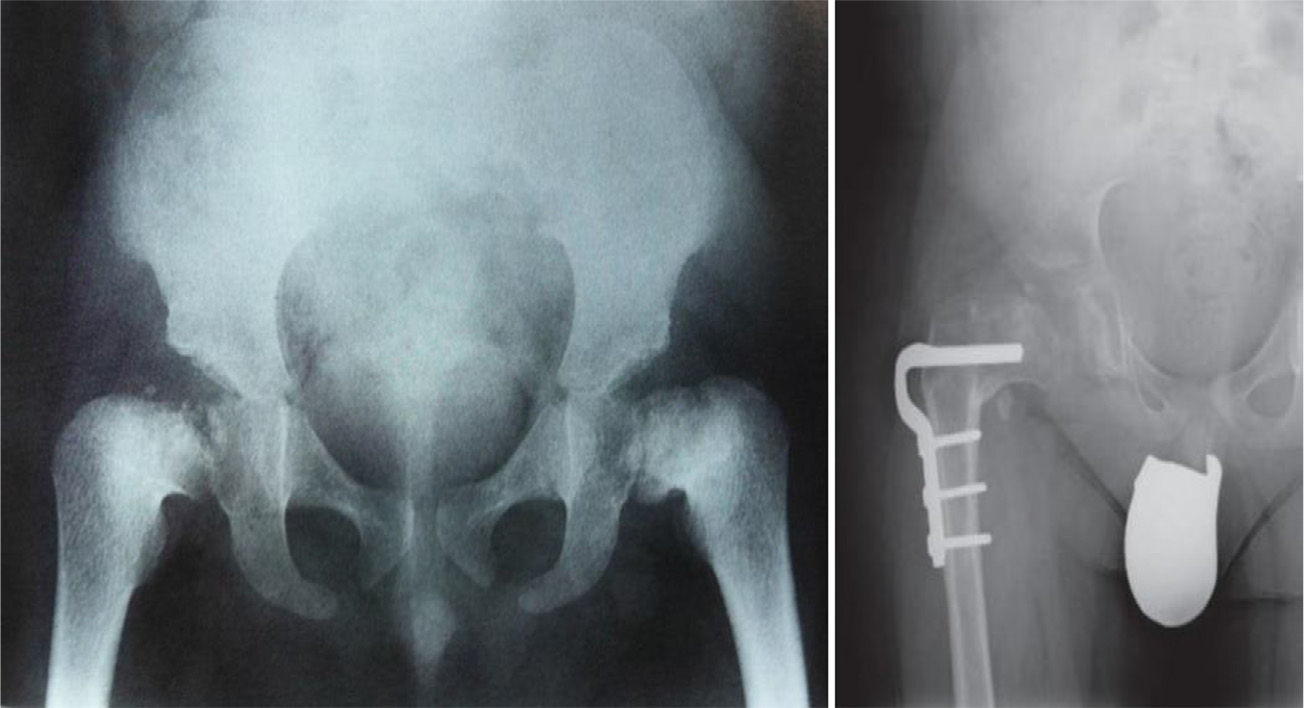
TreatmentTreatment is symptomatic and its key aspects are early diagnosis and monitoring.1–5 Rest and unloading with canes are indicated in coxalgia during childhood. The use of paracetamol is recommended in patients with nocturnal increase of hip pain.1 There is no evidence that femoral osteotomies improve the prognosis or the future need for total hip arthroplasty3 (Fig. 4). In fact, they are even contraindicated in patients with a significant degree of preexisting coxa vara.2 Total bilateral hip arthroplasty at an early age will be the ultimate solution for most patients. In some cases, glenohumeral joint replacement may also be necessary.

Life expectancy is normal and the fundamental limitation is premature coxarthrosis. The final height ranges between 145 and 170cm.
Spondyloepiphyseal dysplasiaCharacteristicsSED includes a group of disorders which are characterized by delayed ossification of the epiphyses of long bones and the axial skeleton. Its prevalence is estimated at 7–11 cases per million inhabitants. Several disorders can de distinguished within this group, such as type II achondrogenesis and hypochondrogenesis, both lethal in newborns, and the congenital and late forms of SED itself.
Congenital spondyloepiphyseal dysplasiaThe reported prevalence is of 3–4 cases per million inhabitants.2 This is an autosomal dominant disorder although most new cases arise from de novo mutations2,3 in the gene encoding type II collagen, located on the long arm of chromosome 12, and generating an impaired function of this protein in the growth and joint plates, intervertebral discs and vitreous humor.1,3 This pathology is evident at birth due to its characteristic phenotype: rhizomelic dwarfism, flattened face with malar hypoplasia and cleft palate, pectus carinatum (or chicken breast) and clubfoot.1–5 Hypotonia and delayed motor development are common during infancy and it is important to rule out that the underlying cause is cervical myelopathy caused by atlantoaxial instability, which is frequent among these patients. Another significant complication in this period is respiratory failure as a result of the small size of the chest cavity.2,4 Lumbar lordosis, development of progressive kyphoscoliosis and waddling gait due to coxa vara and flexion contracture of the hips are common features during the first years of life of these patients. Premature osteoarthritis starts to appear during adolescence and adulthood, especially in the hips and knees. Extraskeletal disorders include a high risk of retinal detachment during adolescence and the possibility of developing hearing problems.1,2,4 Radiological anomalies are present at birth and consist in a delay in virtually all ossification centers,1–5 with the proximal femur and tibia, pelvis and spine being the most affected.1 Common findings at the spinal level include odontoid hypoplasia or the presence of an os odontoideum, narrowing of the intervertebral space, progressive lumbar lordosis and thoracic kyphoscoliosis with a sharp angulation concentrated in several vertebral bodies.2 A lack of ossification of the proximal femoral epiphysis during childhood gives rise to progressive coxa vara, elevation of the greater trochanter and shortening of the femoral neck,1,2 all of which, combined with hip flexion, contribute to premature osteoarthritis. Regarding the management of the disease, the first point is to establish a differential diagnosis with some forms of mucopolysaccharidoses, mainly Morquio disease, with hypothyroidism and pseudoachondroplasia.3 The diagnosis, as well as subsequent follow-up, requires a complete bone map. The most severe complication is cervical myelopathy due to atlantoaxial instability, whose suspicion must lead to a thorough neurological examination, cervical radiographs in flexion and extension and even an MRI scan.1,2 Treatment options include atlantoaxial fusion, surgical decompression and fusion of the atlas to the occiput.2 Regarding scoliosis, present in over half of all patients, if the curve is less than 40° it should attempt to be controlled using a brace,2 with surgical correction being reserved for greater curves. Kyphosis can be optimally controlled using a Milwaukee brace as long as it is maintained until skeletal maturity.2 Virtually all patients will require both femoral and acetabular osteotomies for the treatment of coxa vara, with the combined treatment of flexion contractures also being advisable.2 Pauwel's Y-shaped intertrochanteric osteotomy is particularly indicated in these patients.1 The alignment of the knees should be assessed prior to the correction of the hips, and be corrected in the same surgical procedure, if necessary. In most patients, prosthetic replacement of the affected joints at an early age will be unavoidable,1–5 in many cases requiring new, concomitant osteotomies. Foot deformities will be treated in the same manner as in patients who are not affected by this disease.2 The final size of these patients usually ranges between 80 and 125cm.1,2
Late spondyloepiphyseal dysplasiaThis is an X-linked disorder and therefore more severe in males, although a recessive form has also been described. It is caused by a mutation in type II collagen.2 Clinical manifestations do not appear until infancy, when short stature accompanied by pain in the hips, knees and back becomes evident. The typical features of this disease are shortened trunk, pectus carinatum and premature osteoarthritis, particularly in the hips and shoulders.1–5 The final height is around 150cm. Radiologically it is possible to observe predominant involvement of the shoulders, hips and knees, as well as generalized platyspondylia. Odontoid hypoplasia and also os odontoideum may be present and cause atlantoaxial instability, which must be ruled out,2 as well as scoliosis. Clinical and radiological association is necessary in order to establish the diagnosis of the disease and carry out a differential diagnosis with MED (it rarely affects the axial skeleton) and Scheuermann disease (vertebral involvement is generalized).1 Total prosthetic replacement, preceded or not by corrective osteotomies, will be necessary in most patients at younger ages than in the general population.1–5
Osteogenesis imperfectaOsteogenesis imperfecta (OI) is the most common cause of osteoporosis with a genetic origin.4 It has an estimated incidence of 1:20,000 live newborns,1–5 although a type VIII mutation of the disease has been described in West Africa with an incidence of 1:200–300 among African American carriers.4
It has considerable variability, caused by quantitative and/or structural defects in type I collagen, the main component of the extracellular matrix of bone and skin tissue.4 Most cases follow a pattern of autosomal dominant inheritance, either due to preexisting or de novo mutations, although this condition may also be inherited in an autosomal recessive pattern.1,3,4 Autosomal dominant forms may be caused by 2 types of mutations of the genes which assemble and encode type I collagen: inactivation of one allele of the gene, resulting in a 50% reduction of the amount of collagen, structurally normal, or reduction of the amount of type I collagen associated with a mutation of one of the chains, with a quantitative and structural collagen reduction.1 The result is the synthesis of bones with fine and fibrous trabeculae and with an abnormal proteoglycan and fundamental substance3 content, which are very sensitive to breakages and deformities.
Clinically, the disease is characterized by the triad of Eddowes van der Hoeve: bone fragility, blue sclera and early deafness.4,19 At present, 8 different types of the disease are recognized, based on the 4 types described by Sillence in 19791,4,20,21 according to clinical and radiological criteria:
Type I. The classical form of OI, as well as the most common and mildest.1 It is transmitted in an autosomal dominant pattern and patients present the classic triad. Two subtypes are identified, A and B, depending on the presentation (B) or not (A) of dentinogenesis imperfecta.1,4 Fractures are more frequent than in healthy children and appear when the child begins to walk, decreasing in frequency after puberty. Subsequently, the incidence of fractures increases again in adult age.1,4 Consolidation of bone fractures follows a normal process.
Type II. With 2 subtypes, this is the most severe form of the disease and is lethal prenatally or during the first year of life.1,3,4
Type III. This is the most severe, non-deadly form and the one with greater orthopedic interest.19 Newborns present multiple fractures within the uterus and at birth, with a widened thorax, unlike in type II. Fractures present malunion, contributing to increase the existing skeletal deformities.1,4 Two subtypes can be distinguished: with an autosomal dominant inheritance pattern with mutations in genes encoding type I collagen, and recessive with mutations in genes related to CRTAP.1 Although the weight and length at birth are almost normal, the height reached by these patients is extremely short. It is frequently associated with dentinogenesis imperfecta. Long bones tend to gradually arch, the epiphyses are large compared with the diaphyses and the metaphyses present islands of endochondral ossification with an appearance of “popcorn” in conventional radiology.1,4 In addition, almost all subjects present kyphoscoliosis and progressive vertebral compression. Children are able to walk for a certain period of time, but most stop doing so as they grow.1
Type IV. Similar to type I, although more serious and with normal sclera.1,4 Most subjects do not suffer gross deformities. Ambulation is generally achieved by all subjects and brings new fractures, whose frequency decreases after puberty. Radiologically there is osteoporosis, metaphyseal widening and vertebral compression. The main differential diagnosis of this type is with shaken baby syndrome.1,3–5Type V. Clinically similar to type IV but with different radiological and histological findings.4 It follows an autosomal dominant pattern and its underlying genetic defect is still unknkown.1 Radiographs of the forearms and legs show ossification of the interosseous membranes and radiopaque metaphyseal bands. Subjects present hyperplastic fracture calluses which can even resemble osteosarcoma.1,4
Type VI. Clinically similar to type IV but more severe. Subjects do not present detectable mutations in type I collagen.1 The transmission pattern and the genetic defect are still unknown.
Type VII. Transmitted in an autosomal recessive pattern and often presenting null mutations in P3H1 and CRTAP.4 Subjects present multiple fractures at birth and during childhood whose frequency decreases in adolescence.1 Other characteristics of this type of OI are rhizomelic dwarfism, severe coxa vara and progressive skeletal deformities.
Type VIII. Its transmission pattern and mutation are the same as in type VII and it can be described as an overlap between forms II and III.1,4 Many subjects die during childhood.
DiagnosisIn most cases the diagnosis is established through the symptoms, family history and conventional radiology.1,3–5 Sporadic forms of type IV are the most difficult to diagnose and are often confused with shaken baby syndrome, requiring careful evaluation of radiographs or bone densitometry to dismiss it if doubts persist. Collagen biochemical studies confirm the diagnosis, although they are not performed routinely,1 and DNA sequencing is useful to distinguish between different types and to facilitate prenatal diagnosis and familial studies.4 Other differential diagnoses to take into consideration are3: camptomelic dysplasia, which causes long bones to bow, respiratory failure and numerous extraskeletal deformities, pycnodysostosis, with a greater incidence of fractures but with increased bone density, idiopathic juvenile osteoporosis, in which recurrent fractures are usually metaphyseal rather than diaphyseal and the new bone formed is already osteoporotic rather than normal, with the consolidation process being known as neo-osteoporosis,5 and osteomalacia, which has specific biochemical findings.
TreatmentThere is no cure for the disease. Gene therapy is difficult to apply because each patient carries a different mutation which is responsible for the disease.1 The use of growth hormone treatment has only brought histological benefits.4,19 Bisphosphonates are currently being used as a baseline medical treatment,1,2,4,5,19,21 but their use is not without controversy, its exact indications are not known precisely and the duration of treatment has not been clearly established. It has been demonstrated that they reduce the number of fractures and increase bone volume by decreasing osteoclastic resorption, especially in the case of pamidronate.21–26 More recent publications do not report any doubts regarding the efficacy of bisphosphonates in reducing the number of fractures, increasing bone mass and decreasing pain of skeletal origin in patients with OI,21,27–30 so their indication seems to be widely accepted. The questions, which are currently emerging address the duration of treatment and the side effects derived thereof. Nicolaou et al.31–35 conducted a retrospective study comparing femoral fractures in 176 patients with OI treated with bisphosphonates for 2 years, and observed a decrease in the number of femoral fractures in the group treated with bisphosphonates, along with an increase of fractures in the subtrochanteric region of the femur, many of them due to low-energy trauma, like in osteoporotic patients treated with bisphosphonates. Therefore, they recommended avoiding corrective osteotomies at this level and performing radiographic screening, especially in subjects reporting pain in that area. According to several studies, the association of bisphosphonates with alfacalcidol is more effective in increasing bone mineral density (BMD) and preventing fractures than the isolated use of bisphosphonates.36,37 Iwamoto et al. presented 1 case of an adult patient with osteogenesis imperfecta treated for 11 years with this combination.38 This pattern has been associated with an increase in BMD, an absence of fractures in long bones and a significant improvement in back pain.
Furthermore, most studies have relied on the exclusive use of pamidronate.22–26 Ingmar et al. analyzed ibandronate in 27 patients suffering OI39 and concluded that its use normalizes bone replacement markers and produces an increase in BMD.
New lines of treatment in this disease are bone marrow and mesenchymal stromal cell transplant, and the use of denosumab. The latter is a monoclonal antibody against RANKL, which inhibits the formation of osteoclasts and bone degradation, so it is indicated for the treatment of osteoporosis.40–44 Among the few studies examining the use of denosumab in OI is that by Semler et al.,44 who concluded that the use of the anti-RANKL monoclonal antibody produced a suppression of bone resorption based on the normalization of biochemical markers of bone turnover, a decrease in serum calcium levels without symptoms of hypocalcemia, and a decrease in fracture rate. They also established that drug degradation occurred 3–4 months after administration, unlike the case of bisphosphonates, which remain stored in the skeleton for years, and recommended an administration regime of 2 months for children. For these reasons, the authors encouraged further studies with this monoclonal antibody in patients with OI and left open a new and promising treatment option.
Regarding the treatment of fractures, the consolidation process is totally normal,1,3–5 except in type V in which there are hypertrophic calluses. Therefore, the only differences in the management of fractures are1,4: minimizing immobilization to reduce the osteoporosis it causes, establishing clinical diagnoses of the fractures (reserving radiology for doubtful cases) and employing surgery as conservatively as possible, since patients will require it more than once.
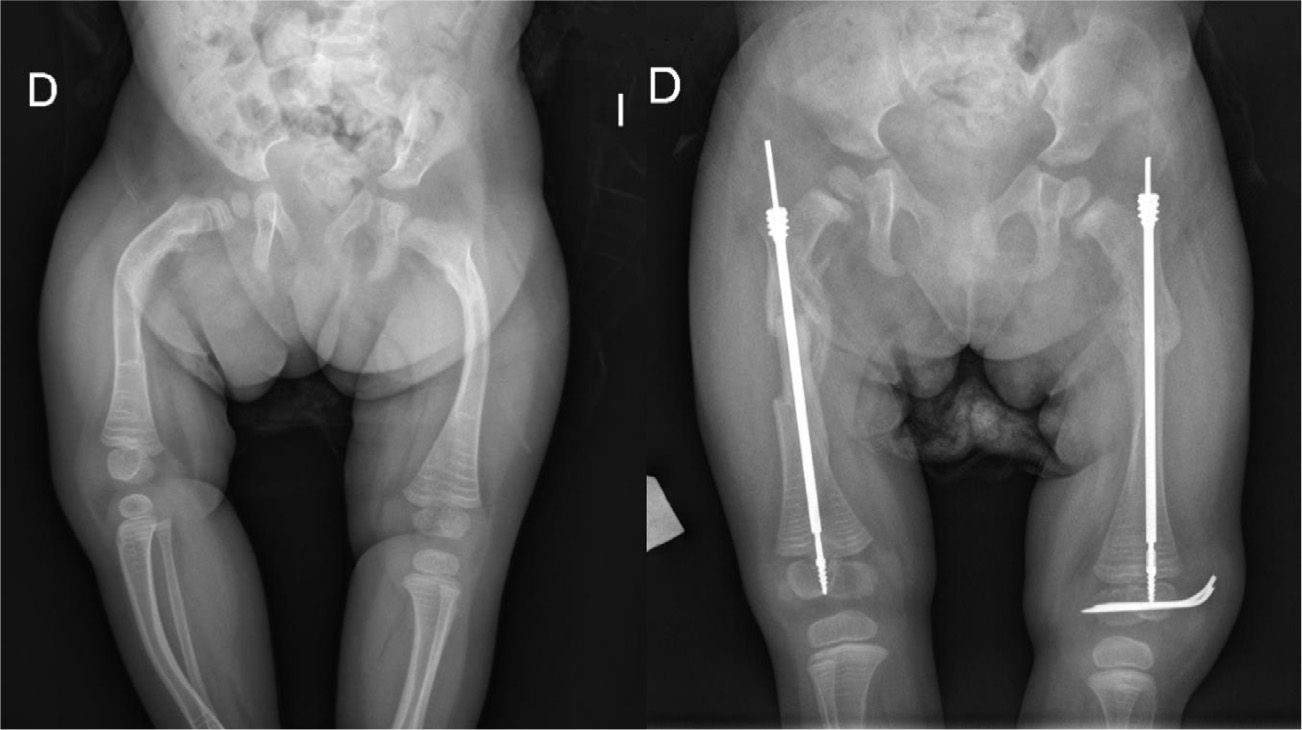
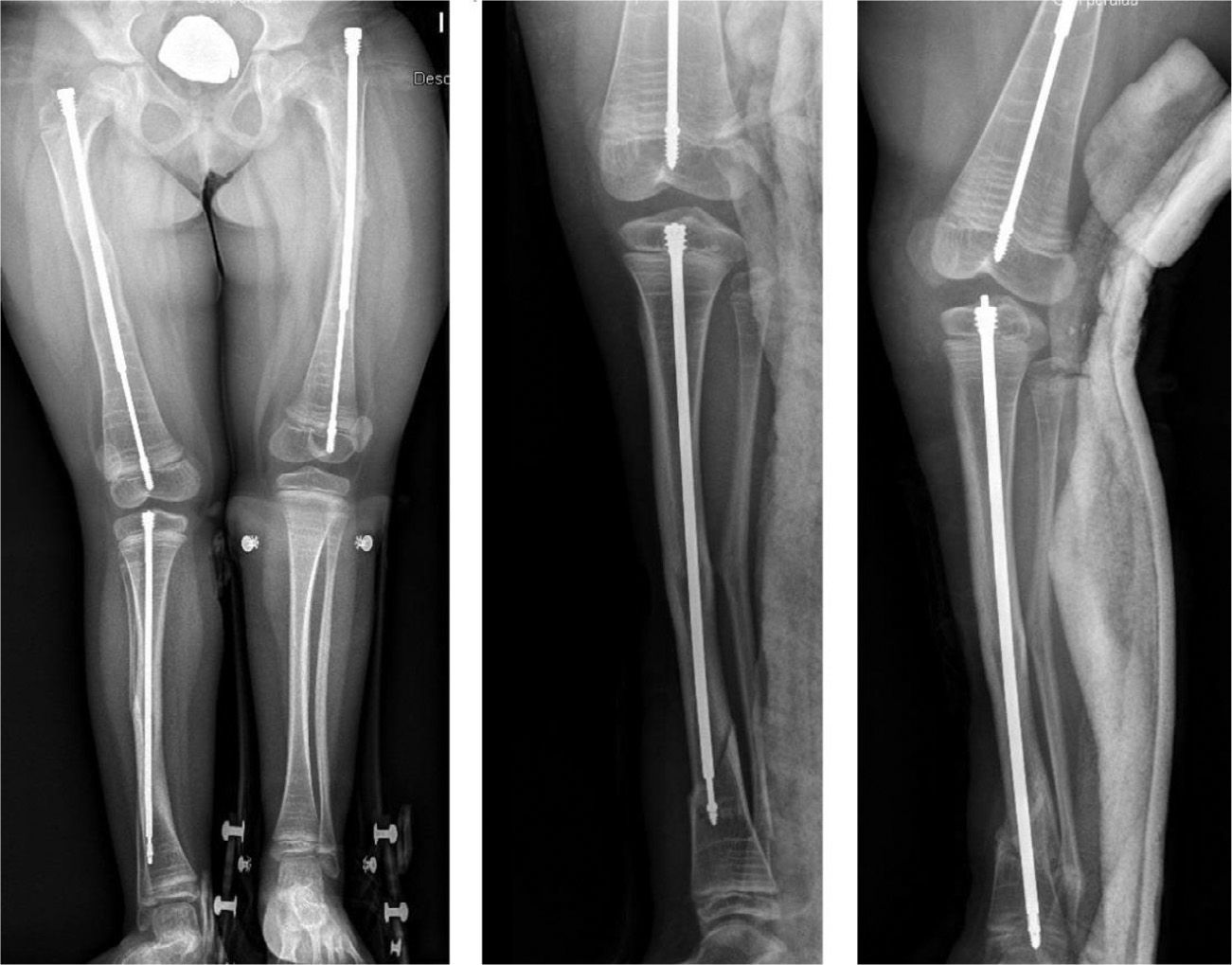
Intramedullary nailing is the treatment of choice for recurrent fractures and long bone deformities.1,3–5 The endomedullary systems employed at present “grow” with the bone (Figs. 5 and 6) allowing them to remain in position for long periods, serving as “guides” to avoid the appearance of new fractures.


Skeletal dysplasias are a major cause of severe growth development. For daily clinical practice it is more useful to classify them according to the affected area of the skeleton: spondylodysplasias, metaphyseal dysplasias, epiphyseal dysplasias and combinations of the above. Achondroplasia is the most common.
In achondroplasia it is important to rule out the symptoms arising from foramen magnum and vertebral canal stenosis, which despite not representing the most common complication is the most serious. Bone elongation is still a controversial treatment option. The latest lines of research have focused on biological drugs which reduce the activity of FGFR3.
MED is currently considered as a disease with variable expressivity. The main differential diagnosis is with Perthes disease.
SED has 2 forms, congenital and late, and both display axial and epiphyseal involvement, although with varying degrees. Atlantoaxial instability is a common and significant anomaly in the diagnosis and monitoring of patients, as it can cause cervical myelopathy.
In OI the process of fracture consolidation is normal, so its treatment is similar to that of fractures occurring in healthy children, avoiding prolonged immobilization because it increases osteoporosis. A total of 8 different types of OI can be distinguished, with type III being most serious non-fatal type and, therefore, the one with most orthopedic interest, whilst type IV is the one with greater legal implications, as it can be mistaken for shaken baby syndrome. Bisphosphonates are currently being used for primary treatment of the disease; the latest lines of research have focused on the use of denosumab, which has offered encouraging results.
Level of evidenceLevel of evidence v.
Ethical responsibilitiesProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Borrego E, Farrington DM, Downey FJ. Novedades en displasias óseas. Rev Esp Cir Ortop Traumatol. 2014;58:171–181.
