



La dermatomiositis clínicamente amiopática comprende un grupo especial de pacientes dentro del espectro de la dermatomiositis, caracterizados por la presencia de lesiones cutáneas típicas, compromiso muscular mínimo o ausente y riesgo aumentado de enfermedad pulmonar intersticial. Los anticuerpos dirigidos contra la proteína codificada por el gen asociado con la diferenciación del melanoma 5 (MDA5), están presentes en una proporción importante de pacientes con dermatomiositis clínicamente amiopática, los cuales desarrollan enfermedad pulmonar intersticial rápidamente progresiva, con elevada mortalidad y que se complica frecuentemente con la aparición de neumomediastino espontáneo. Presentamos el caso de una paciente de origen africano con dermatomiositis clínicamente amiopática anti-MDA5 positiva y enfermedad pulmonar intersticial con patrón tomográfico de neumonía organizada, que desarrolló neumomediastino espontáneo durante su evolución.
Clinically amyopathic dermatomyositis comprises a special group of patients within the spectrum of dermatomyositis characterised by the presence of typical skin lesions, minimal or absent muscle involvement, and increased risk of interstitial lung disease. The antibodies directed against the protein encoded by melanoma differentiation-associated gene 5 (MDA5) are present in a significant proportion of patients with clinically amyopathic dermatomyositis, who develop rapidly progressive interstitial lung disease, with high mortality and frequently complicated by the onset of spontaneous pneumomediastinum. A case is presented of an African patient with anti-MDA5 positive clinically amyopathic dermatomyositis and interstitial lung disease with tomography pattern of organising pneumonia who developed spontaneous pneumomediastinum during its clinical course.
La dermatomiositis (DM) es una enfermedad inflamatoria sistémica que se caracteriza por debilidad muscular proximal, mialgias y manifestaciones cutáneas típicas como el eritema «en heliotropo» y las pápulas de Gottron. Sin embargo, algunos pacientes con DM no desarrollan alteraciones musculares (DM amiopática), mientras que otros presentan síntomas leves durante el curso de la enfermedad o el compromiso muscular solo se evidencia por elevación de las enzimas asociadas con daño muscular o alteraciones miopáticas en el electromiograma o la biopsia muscular (DM hipomiopática), por lo que se incluyen dentro del espectro de la DM clínicamente amiopática (DMCA), teniendo este grupo un riesgo aumentado de desarrollar enfermedad pulmonar intersticial (EPI)1.
Aunque su etiología es desconocida, existe evidencia de que el daño tisular en la DM es producido por mecanismos autoinmunes, encontrándose autoanticuerpos circulantes específicos de miositis como anti-Mi2 y anti-Jo1 en 50-70% de los casos2. Recientemente se han identificado anticuerpos dirigidos contra la proteína codificada por el gen asociado con la diferenciación del melanoma 5 (melanoma differentiation-associated gene 5 [MDA5], perteneciente a la familia de receptores Rig-I-like, que se relacionan con la respuesta a infecciones virales), los cuales están presentes en 19-35% de los pacientes con DMCA y se asocian específicamente con compromiso muscular mínimo o ausente y EPI rápidamente progresiva, complicada frecuentemente por la aparición de neumomediastino espontáneo (NE)3,4. Presentamos el caso de una paciente de origen africano con DMCA y positividad para anti-MDA5 que desarrolló EPI y NE durante su evolución.
Presentación de casoMujer de 18 años de raza negra, natural de Sahara Occidental, comenzó a presentar a los 14 años poliartritis simétrica, fiebre y alopecia. Fue trasladada a Argelia a los 16 años donde se inició tratamieno con prednisona oral (dosis máxima 10mg/día), desarrollando poco después fenómeno de Raynaud y ulceraciones en pulpejos de dedos de ambas manos. Fue ingresada a los 17 años por disnea y tos no productiva, evidenciándose opacidades bibasales y espirometría alterada con patrón restrictivo severo (capacidad vital forzada 33% de su valor teórico), recibiendo antiobioticoterapia intravenosa empírica y siendo dada de alta con mejoría parcial. Durante dicho ingreso se detectó factor reumatoide (FR) positivo (108 UI/ml), por lo que fue diagnosticada de artritis reumatoide y se inició administración de metotrexato oral (10mg/semana) más suplemento de ácido fólico (5mg/semana).
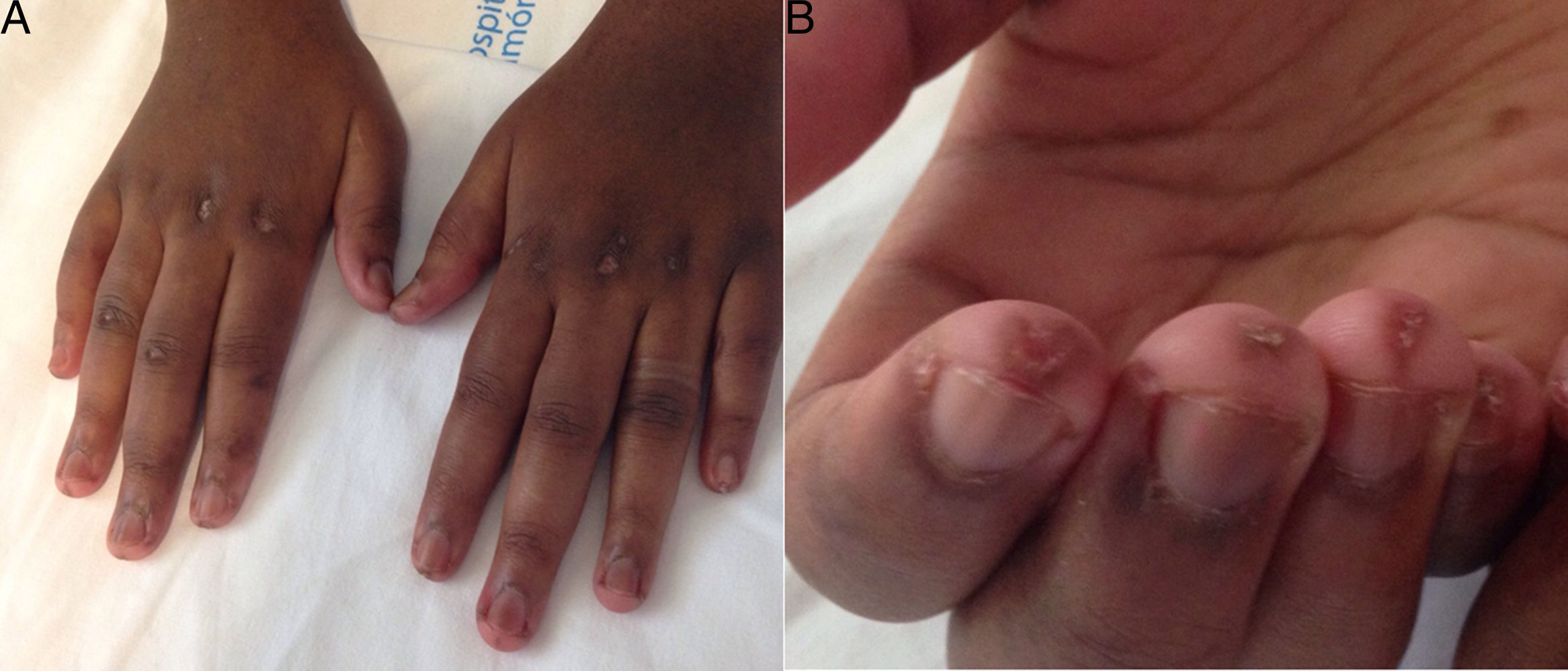
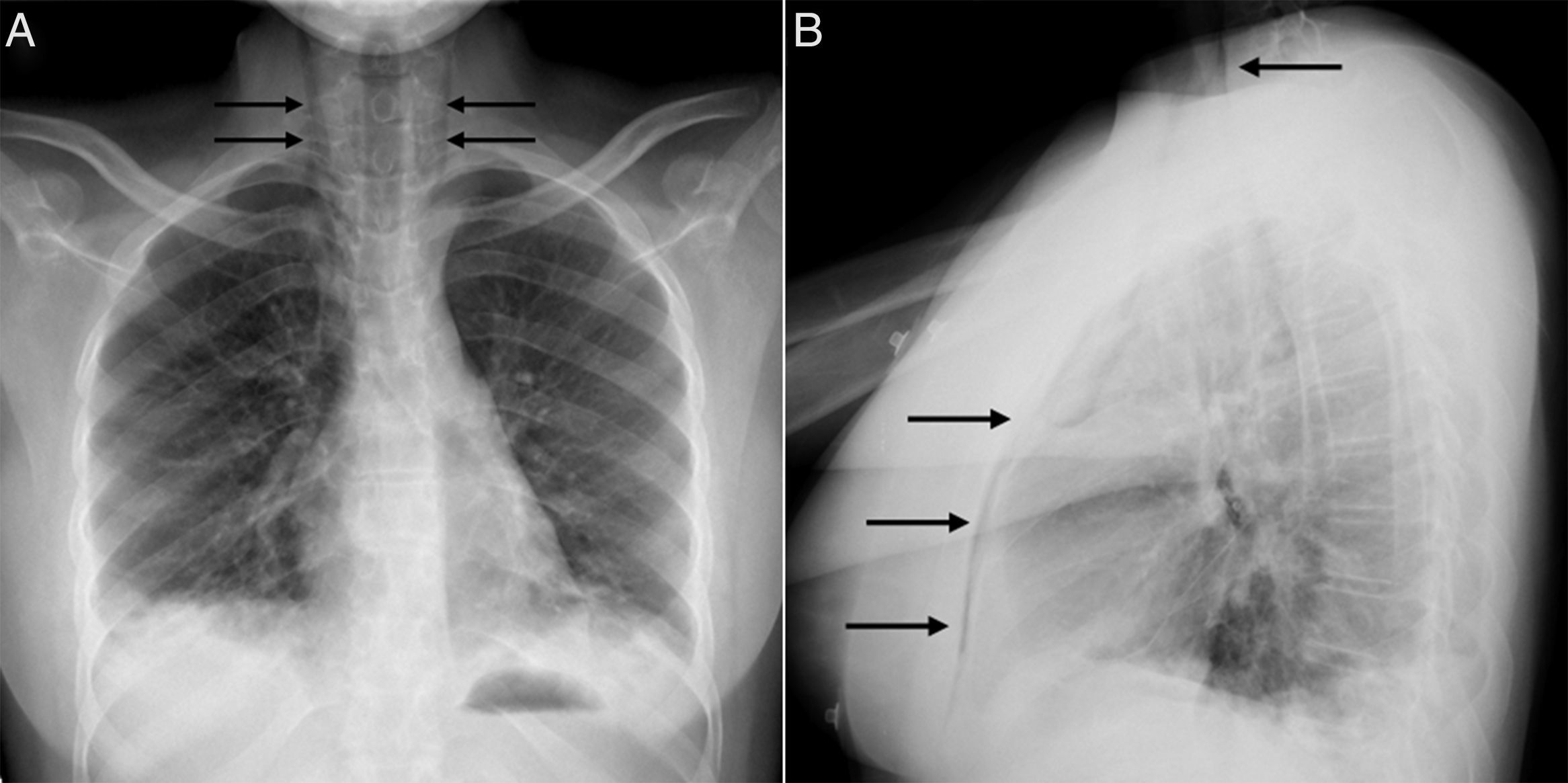
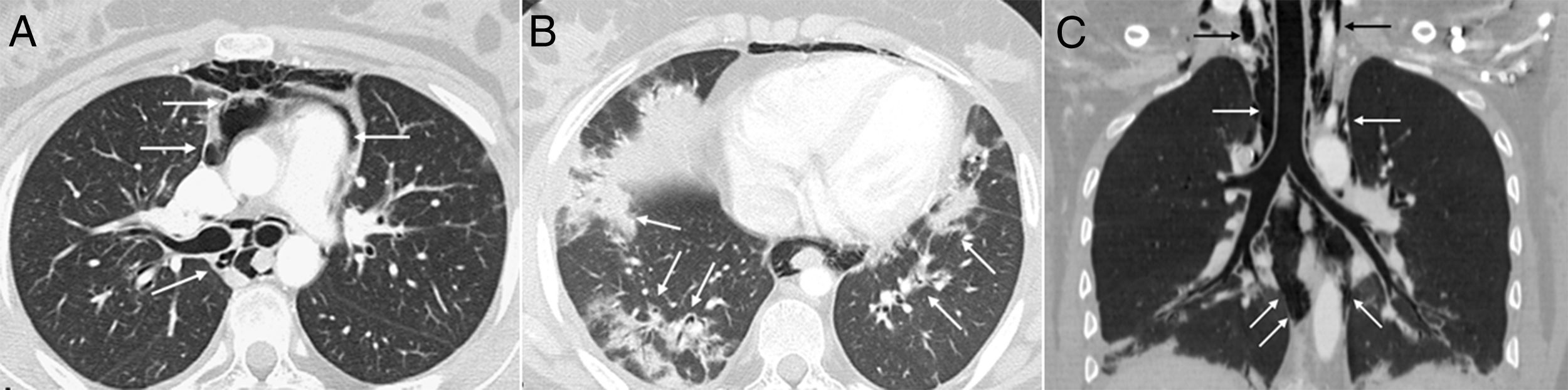
A los 18 años fue trasladada a España e ingresó en nuestro centro, presentando en este momento edema difuso de manos, lesiones cicatriciales en los pulpejos, eritema periungueal, máculas hipopigmentadas de aspecto atrófico sobre zonas en las que previamente tuvo pápulas de Gottron en metacarpofalángicas e interfalángicas proximales (fig. 1), nódulos subcutáneos indurados en antebrazos y muslos, disnea de esfuerzos leves, disfagia, pirosis y astenia, pero no debilidad muscular, ni mialgias. La saturación de oxígeno basal era normal (97%) pero persistía con patrón restrictivo (capacidad vital forzada 34,8%), no pudiendo realizarse la prueba de capacidad de difusión de monóxido de carbono (DLCO) porque movilizaba un volumen insuficiente. La analítica mostraba anemia microcítica hipocrómica (hemoglobina 11,8g/dl, volumen corpuscular medio 74,6 fl, hemoglobina corpuscular media 23,7 pg), linfopenia (750/mm3), aumento de reactantes de fase aguda (velocidad de eritrosedimentación 84mm/h, proteína C reactiva 17mg/l), hipergammaglobulinemia policlonal (IgG 2800mg/dl) y aumento de hormona estimulante de la tiroides (TSH) (5,230μUI/ml) con niveles de T3 y T4 normales. El resto del estudio bioquímico incluyendo enzimas musculares (creatinquinasa 40 U/l, aldolasa 2 U/l) estaba dentro de rangos normales. Desde el punto de vista inmunológico presentaba ANA 1/80 con patrón nucleolar, anti-Ro52 y anti-MDA5 positivos, siendo negativos los demás anticuerpos estudiados (FR, antipéptido cíclico citrulinado [anti-PCC], anti-DNA, anti-ENA, anti-Scl70, anticentrómero, anticuerpos antifosfolípidos, anti-Mi2, anti-Jo1, anti-PL12, anti-PL7, anti-OJ, anti-EJ, anti-SRP, anti-Ku y anti-PM/Scl). La radiografía de tórax mostraba las opacidades bibasales y signos de NE que se extendían hasta la región peritiroidea (fig. 2), aunque en la exploración física no se evidenciaba crepitación en tejido celular subcutáneo, por lo que se realizó una tomografía computarizada que confirmó la presencia de opacidades parcheadas peribronquiales de predominio bibasal, compatibles radiológicamente con un patrón de neumonía organizada y un extenso neumomediastino que se extendía desde la región tiroidea a lo largo de todo el mediastino disecando estructuras vasculares y musculares (fig. 3). No se apreciaban signos de panalización subpleural, mediastinitis o neumotórax.
. En la cara palmar se evidencian lesiones cicatriciales en pulpejos de dedos B).")
Aspecto dorsal de las manos de la paciente mostrando edema difuso hasta falanges medias, máculas hipopigmentadas de aspecto atrófico en metacarpofalángicas e interfalángicas proximales y eritema periungueal A). En la cara palmar se evidencian lesiones cicatriciales en pulpejos de dedos B).
, sin neumotórax ni enfisema subcutáneo asociados, y opacidades parenquimatosas bibasales A) y B).")
 A). Otra imagen axial demuestra la existencia de opacidades parcheadas peribronquiales en ambas bases pulmonares, compatibles con un patrón de neumonía organizada (flechas) B). En la reconstrucción coronal se observa un extenso neumomediastino que se extiende desde la región tiroidea (flechas negras) a lo largo de todo el mediastino (flechas blancas) C).")
Imagen axial de TC de tórax en la que se confirma la presencia de gas disecando las estructuras del mediastino (flechas) A). Otra imagen axial demuestra la existencia de opacidades parcheadas peribronquiales en ambas bases pulmonares, compatibles con un patrón de neumonía organizada (flechas) B). En la reconstrucción coronal se observa un extenso neumomediastino que se extiende desde la región tiroidea (flechas negras) a lo largo de todo el mediastino (flechas blancas) C).
La paciente fue diagnosticada inicialmente de DM amiopática con EPI y NE asociados, pero el electromiograma demostró actividad espontánea sugestiva de miopatía inflamatoria leve y en la biopsia muscular se encontraron cambios miopáticos inespecíficos sin infiltrado inflamatorio, por lo que correspondía en realidad a DM hipomiopática. Se completó el estudio con capilaroscopia que evidenciaba megacapilares aislados con densidad capilar conservada, manometría esofágica con contracciones débiles a nivel de mitad inferior de esófago y biopsia de los nódulos subcutáneos con hallazgos compatibles con paniculitis inespecífica. Se inició administración de prednisona 1mg/kg/día con mejoría rápida de la disnea, lesiones cutáneas y astenia. Se suspendió el metotrexato y se añadió azatioprina 100mg/día. En la revisión a los 2 meses del alta la paciente se encontraba estable, con disnea leve y artralgias sin signos de sinovitis. La radiografía de tórax mostraba una reducción importante de los infiltrados pulmonares y desaparición de los signos de NE. La paciente abandonó el seguimiento por traslado domiciliario.
DiscusiónLos anticuerpos anti-MDA5 (anteriormente llamados anti-CADM140) fueron descritos en el año 2005 por Sato et al., en pacientes japoneses con DMCA y EPI rápidamente progresiva5. Estudios posteriores han encontrado estos anticuerpos en otras poblaciones y ampliado su espectro de manifestaciones clínicas, siendo característica la presencia de ulceraciones cutáneas y orales, pápulas/pústulas palmares dolorosas, eritema periungueal, eritema en codos/rodillas (signo de Gottron), alopecia difusa, paniculitis y artralgias/artritis3,4,6. Debido a estas características, así como al escaso compromiso muscular, la preponderancia de la EPI y la infrecuente asociación con neoplasias, se ha propuesto denominar a este cuadro como «síndrome dermatopulmonar asociado a MDA5» para diferenciarlo de la DM clásica6, aunque estos anticuerpos no son exclusivos de la DMCA y también han sido descritos en las formas clásica y juvenil de la DM7. La capacidad de los anticuerpos anti-MDA5 para identificar pacientes con DM y riesgo de desarrollar EPI rápidamente progresiva es alta, siendo su sensibilidad del 77% y especificidad del 86%7. El factor racial puede influir la expresividad clínica en pacientes con anti-MDA5. Aproximadamente la mitad de los pacientes provenientes del este asiático (Japón, China y Corea) presentan EPI rápidamente progresiva y generalmente corresponden a DMCA8, mientras que en poblaciones caucásicas la severidad de la EPI puede ser menor y la frecuencia de miositis clínica mayor9. Se ha descrito la DMCA asociada con anti-MDA5 en pacientes afroamericanas9,10, pero según la revisión de la literatura que hemos realizado este es el primer caso en una mujer de origen africano.
En nuestro caso se planteó inicialmente el diagnóstico de AR por la presencia de artritis y FR positivo, un hecho que no es raro en pacientes anti-MDA5 positivos ya que una proporción importante (65,5-81,8%) desarrolla poliartritis simétrica con compromiso de articulaciones pequeñas de manos y rigidez matutina que es indistinguible de la AR, pudiendo algunos de ellos presentar positividad para FR o anti-PCC y, más raramente, erosiones3,9,11. No es tampoco infrecuente el desarrollo de artritis asociada con «manos de mecánico», fenómeno de Raynaud o fiebre en pacientes con positividad para anti-MDA5, lo que también puede originar la sospecha de un síndrome antisintetasa (SAS), recomendándose la determinación de estos anticuerpos cuando los antisintetasa son negativos9. Nuestra paciente presentaba además anticuerpos anti-Ro52, los cuales han sido descritos como coestimuladores en el síndrome antisintetasa, aumentando la gravedad de la ILD en estos casos12,13. Esto también ha sido reportado en pacientes con DMCA anti-MDA5 positiva, encontrándose la presencia simultánea de anti-Ro52 en 19-50% de los casos3,9,14. La coexistencia de ambos anticuerpos probablemente tiene implicaciones en la patogenia de la EPI en este subgrupo de la DM. MDA5 y Ro52 (TRIM21) son proteínas citoplasmáticas cuya expresión es inducida por interferones tipo I, por lo que se ha sugerido que la interacción de ambas moléculas podría conducir a la formación de complejos moleculares con una inmunogenicidad aumentada9.
Una característica peculiar de la EPI asociada a la DM es la frecuente aparición de NE, con una incidencia que varía del 2,2 a 8,3%, correspondiendo más de la mitad de estos casos a DMCA15,16. Se han descrito como factores de riesgo para desarrollar NE la presencia de EPI, vasculopatía cutánea, uso de glucocorticoides sistémicos, paciente joven y niveles séricos normales de enzimas musculares15, todos ellos presentes en nuestro caso. La presencia de anti-MDA5 también parece ser un factor predictor del desarrollo de NE en estos pacientes, tal como lo demuestra el estudio de Koga et al., que incluyó 79 pacientes con DM (58 clásica y 21 amiopática), encontrando que la presencia de enfisema mediastínico fue significativamente mayor en pacientes anti-MDA5 positivos (35 vs. 2%, p=2,1x10-5)4. El mecanismo por el cual se desarrolla el NE en la DM no está claramente establecido, pero se especula que puede ser el resultado de la rotura de bullas o quistes subpleurales secundarios a la fibrosis intersticial y al aumento de la presión intraalveolar. También se ha descrito un debilitamiento de las paredes alveolares debido al tratamiento con glucocorticoides17,18.
Respecto a los patrones tomográficos de EPI encontrados en pacientes anti-MDA5 positivos, el más frecuente es el de consolidación/áreas en vidrio deslustrado basales (50%), seguido por áreas en vidrio deslustrado de distribución aleatoria (33%), mientras que los patrones reticular basal y de consolidación peribroncovascular sugestivo de neumonía organizada suelen ser infrecuentes19. La histopatología de la EPI asociada con DMCA anti-MDA5 positiva no ha sido evaluada sistemáticamente, pero en los reportes de casos con biopsia pulmonar o autopsia, los hallazgos fueron compatibles con daño alveolar difuso, la variante fibrótica de la neumonía intersticial no específica y la neumonía intersticial usual6,14,18-24.
La mayoría de los pacientes con EPI asociada a DM tienen una respuesta favorable al tratamiento con glucocorticoides a dosis altas e inmunosupresores. Sin embargo, aquellos con DMCA y EPI rápidamente progresiva son resistentes a múltiples tratamientos y tienen mal pronóstico, con una mortalidad del 41% en pacientes con anti-MDA5 positivos, por lo que se recomienda tratar de forma temprana y agresiva con combinaciones de fármacos incluyendo glucocorticoides, inhibidores de la calcineurina, micofenolato de mofetilo, inmunoglobulinas intravenosas, ciclofosfamida y rituximab4,8,25. La presencia de NE también influye en el pronóstico, encontrándose una tasa de mortalidad del 34% en pacientes con DM que presentan esta complicación, falleciendo un 25% de ellos durante el primer mes a causa de distrés respiratorio16. Se han publicado 2 casos de EPI rápidamente progresiva en DMCA asociada con anti-MDA5 que fueron refractarios al tratamiento con pulsos de metilprednisolona e inhibidores de la calcineurina y finalmente fueron sometidos a hemoperfusión con polimixina B, una técnica inicialmente desarrollada para remover la endotoxina de la circulación en pacientes con sepsis, que ha demostrado ser eficaz en pacientes con distrés respiratorio. Uno de ellos presentó una mejoría dramática y reducción de los niveles de anticuerpos anti-MDA526, pero el otro caso (que presentaba como complicación NE y enfisema subcutáneo) solo tuvo una mejoría ligera transitoria y requirió plasmaféresis e inmunoglobulinas intravenosas27.
En conclusión, los anticuerpos anti-MDA5 identifican un grupo especial de pacientes con DM que tienen tendencia a presentar formas amiopáticas/hipomiopáticas, con un espectro de manifestaciones mucocutáneas y articulares y un riesgo incrementado de desarrollar EPI, la cual puede seguir un curso rápidamente progresivo y complicarse por la aparición de NE.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
