




La leucodistofia metacromática (LDM) es una enfermedad desmielinizante rara (prevalencia, 1:40.000), también llamada deficiencia de arilsulfatasa A (ARS-A), que puede presentarse con síntomas neurológicos y psiquiátricos y cuyo diagnóstico puede plantear dificultades para el clínico, dado lo inespecífico de los signos y síntomas. Se presenta el caso de una paciente de 16 años atendida por psiquiatría por cambios conductuales, psicosis y deterioro general del funcionamiento. Inicialmente diagnosticada como esquizofrenia, se documentaron por resonancia magnética y pruebas de laboratorio en la evolución cambios que llevaron al diagnóstico de leucodistrofia metacromática.
Metachromatic leukodystrophy (MLD) is a rare demyelinating disease (prevalence 1:40 000), also called arylsulfatase A deficiency (ARS-A), which may present with neurological and psychiatric symptoms. Clinical assessment may be difficult, due to unspecific signs and symptoms. A case is presented of a 16 year-old female patient seen in psychiatry due to behavioural changes, psychosis, and with impaired overall performance. She was initially diagnosed with schizophrenia, but the Nuclear Magnetic Resonance (NMR) scan and laboratory tests lead to the diagnosis of MLD.
La leucodistrofia metacromática (LDM) es un trastorno genético desmielinizante que clínicamente puede cursar con síntomas neuropsiquiátricos similares a los de los pacientes con esquizofrenia, como la psicosis1,2. El inicio tardío de la LDM se ha propuesto como un modelo de esquizofrenia, debido a que ambos trastornos se caracterizan por signos de desconexión anatómica generalizada y alteración funcional secundaria que evidencia una lesión extensa en los circuitos frontales subcorticales, los cuales se manifiestan en síntomas del estado de ánimo, motivación, alteración en el comportamiento, juicio y planeación1, que hace pensar en el concepto de diasquisis acuñado por Monakow (1914), resurgido recientemente, en la cual se aprecian trastornos neurofisiológicos distantes de la lesión focal cerebral3.
Actualmente, gran parte de los pacientes no se someten a la secuenciación de gen ARS-A, aproximadamente el 50% de los alelos no han sido identificados y no es posible predecir la evolución clínica basándose únicamente en el análisis de las mutaciones4.
Presentación de casoLa paciente inició a los 16 años cambios de comportamiento caracterizados por marcado aislamiento, mal desempeño y problemas de conducta en la escuela, alteración del patrón de sueño, con síntomas psicóticos: soliloquios, alucinaciones, comportamientos desorganizados (come plátanos con cáscara) y deterioro general de su funcionamiento, sin recuperación. Se solicitó inicialmente tomografía computarizada (TC) craneal simple, electroencefalograma, hemograma y perfil tiroideo y metabólico, que se informó normales. Se diagnosticó esquizofrenia paranoide. Requirió varias hospitalizaciones en unidad de salud mental e institucionalización en hogar de cuidados crónicos por síntomas psicóticos y graves episodios de agresión a terceros.
Ante la pobre respuesta farmacológica con persistencia de síntomas, se decidió solicitar una resonancia magnética (RM), que mostró pequeñas lesiones focales, inespecíficas, en la sustancia blanca de ambos hemisferios cerebrales, y en el diagnóstico diferencial se incluyó la posibilidad de lesiones isquémicas en territorios de pequeños vasos. La angiografía cerebral resultó normal y el perfil metabólico y los anticuerpos de leptospira, citomegalovirus y virus de Epstein-Barr se reportaron negativos.
Recibió tratamiento con haloperidol, y se presentaron efectos extrapiramidales. Posteriormente, con olanzapina presentó un marcado aumento de peso; con risperidona, amenorrea, y finalmente se la trató con aripiprazol en dosis progresivamente mayores hasta 60 mg, con lo que se logró la estabilización de los síntomas conductuales y psicóticos.
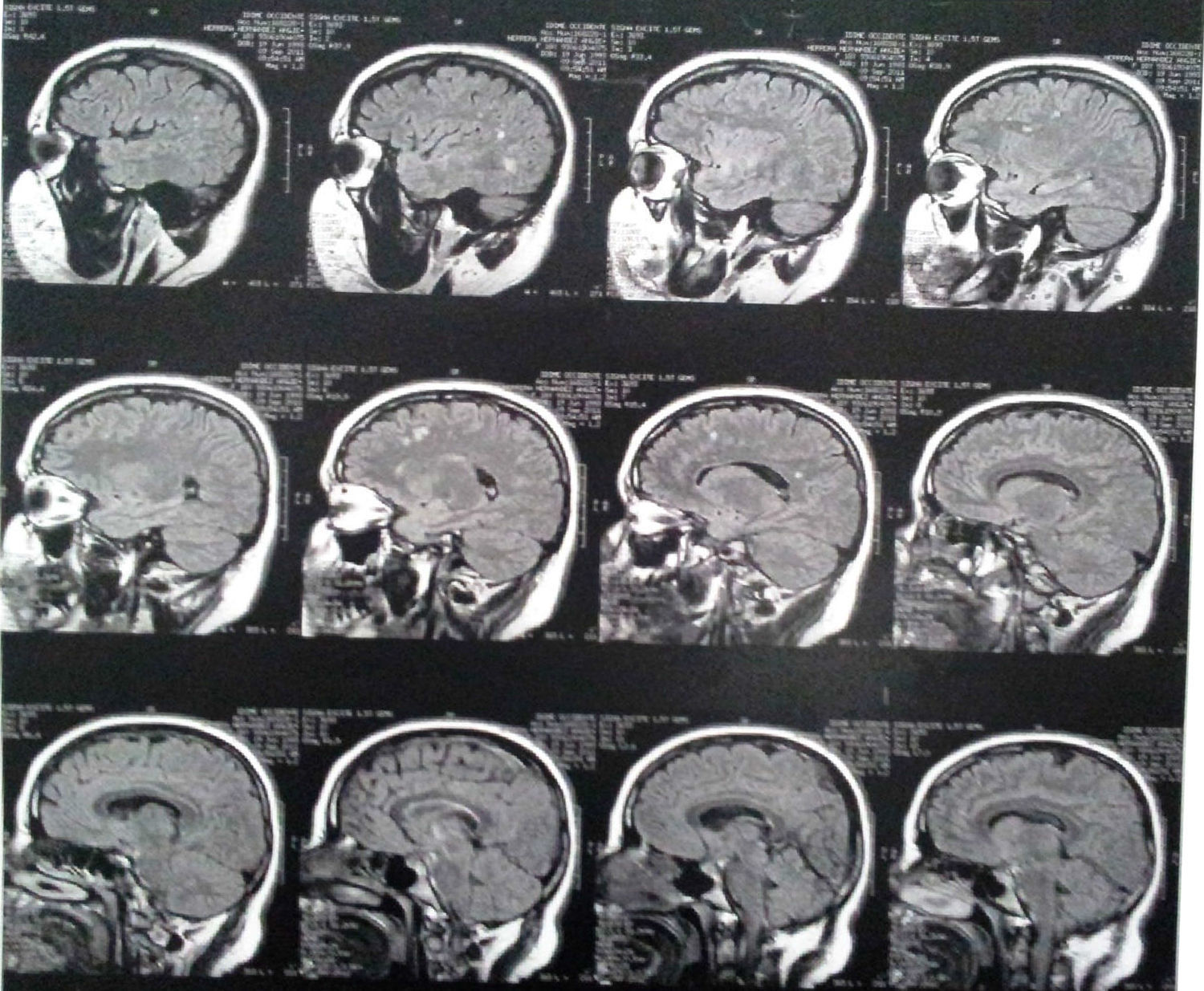
Tres años después de la primera evolución, se apreciaban lesiones focales hiperintensas en T2 y FLAIR frontoparietales subcorticales bilaterales, de carácter inespecífico, más probablemente como secuelas (fig. 1).

El hemograma y las determinaciones de tirotropina, VDRL, función renal y ácido fólico se reportaron normales; los anticuerpos antinucleares y anticardiolipínicos y el perfil completo de fosfolípidos se informaron negativos. Reumatología descartó enfermedad autoinmunitaria como causa del trastorno del comportamiento.
La valoración genética reveló enfermedad de carácter neurodegenerativo y trastorno de la sustancia blanca. Llama la atención el reporte de ácido láctico elevado, y se solicitó cribado metabólico para enfermedad de sustancia blanca. Los resultados reportan disminución importante de ARS-A en leucocitos que, junto con el cuadro clínico y la RM cerebral, llevó al diagnóstico de LDM.
DefiniciónLa LDM es una enfermedad lisosomal del grupo de las esfingoliposis, producida por una deficiencia de ARS-A, una enzima relacionada con el metabolismo de los sulfatos, abundante en la mielina (tabla 1)3. La baja concentración de esta enzima afecta al metabolismo del cerebrósido sulfato y causa la acumulación intralisosomal del sulfátido esfingolípido 3′O-sulfogalactosilceramida en la sustancia blanca del sistema nervioso central (oligodendrocitos y glía) y periférico, especialmente en las vainas de mielina que rodean las células nerviosas y otros tejidos del organismo, como el lactosilsulfátido de los riñones, la vejiga y la vesícula biliar1,5,6. La acumulación de este material altera la formación y degenera la mielina por un mecanismo fisiopatológico desconocido7.
Clasificación de la motricidad gruesa y la función del lenguaje expresivo en la leucodistrofia metacromática15
| Motricidad gruesa (GMFC-MLD) | |
| M0 | Camina sin apoyo, con calidad y desempeño normales para la edad |
| M1 | Camina sin apoyo, pero con calidad y desempeño reducidos; por ejemplo, inestabilidad al permanecer de pie o caminar |
| M2 | Camina con apoyo, caminar sin apoyo no es posible (menos de cinco pasos) |
| M3 | Se sienta sin apoyo, y no es posible la locomoción como rodar o arrastrarse y caminar con o sin apoyo |
| M4 | Se sienta con apoyo, pero no es posible la locomoción o sentarse sin apoyo, pero hay locomoción tal como rodar o arrastrarse |
| M5 | Ni locomoción ni sentarse sin apoyo, pero puede controlar la cabeza |
| M6 | Pérdida de cualquier locomoción y pérdida de control de cabeza y tronco |
| Lenguaje expresivo (ELFC-MLD) | |
| E0 | Se comunica con frases completas de calidad y desempeño normales para la edad |
| E1 | Se comunica con frases completas con calidad y desempeño reducidos para la edad |
| E2 | No puede comunicar frases completas, pero es capaz de usar dos frases cortas |
| E3 | No puede comunicar dos frases cortas, pero es capaz de usar palabras/ideas únicas |
| E4 | Pérdida completa del lenguaje expresivo |
La LDM se debe a una mutación autosómica recesiva del cromosoma 22q, que resulta en una deficiencia de ARS-A. Se han identificado 189 mutaciones en el gen ARS-A8, incluida la sustitución en el aminoácido 31, una sustitución en la posición, tres deleciones, tres mutaciones en sitio de donante y tres mutaciones en sitio de unión del receptor con el donante1.
Salmon et al. reportaron el caso de una mujer de 30 años con diagnóstico de LDM confirmada enzimáticamente, con cambios cognitivos en los que se evidenciaba hipometabolismo bilateral en el tálamo, la corteza frontal medial, el polo frontal y la corteza occipital, a diferencia de los cambios mostrados en el Alzheimer: hipoperfusión en región frontal dorsolateral y temporal1.
Tamagakien informó de cambios similares en un paciente con cambios de conducta1.
DiagnósticoEl diagnóstico se sospecha cuando se encuentran gránulos metalocromáticos en biopsia de la conjuntiva o nervio sural1. El diagnóstico se confirma cuando se documentan escasa actividad de ARS-A en leucocitos o cultivo de fibroblastos1,9.
En la seudodeficiencia de ARS-A, hay un déficit parcial que no causa trastornos clínicos, lo cual puede complicar el diagnóstico y la identificación de quienes padecen LDM. Esta condición también se puede encontrar en sujetos sanos4.
El diagnóstico radica en análisis de las mutaciones, pruebas bioquímicas y evaluaciones clínicas4.
Los exámenes que se puede usar para el diagnóstico de LDM incluyen10:
- •
Análisis de sangre o piel para detectar si hay poca actividad de ARS-A.
- •
RM cerebral.
- •
Punción lumbar para evaluar si hay altas concentraciones de proteína.
- •
Análisis de orina para ver si hay altas concentraciones de sulfátidos.
- •
Estudios de velocidad de la conducción nerviosa.
El sulfátido en nervio y líquido cefalorraquídeo y la acumulación de lisosulfátido proveen un marcador de la gravedad en nervio periférico únicamente; no refleja la extensión de la lesión en el sistema nervioso central11.
Diagnóstico prenatal y cribado del estado de portadorAntes del nacimiento, se puede determinar la actividad de la ARS-A mediante cultivo celular de líquido amniótico o de vellosidades coriónicas. El diagnóstico prenatal se indica a las parejas que tienen el antecedente de un hijo afectado. El estado de portador se puede reconocer mediendo la actividad de la ARS-A, aunque existen valores que se pueden encontrar en población sana. Es indispensable diferenciar al portador de LDM de la condición benigna; para ello lo más efectivo es el análisis de las mutaciones4.
Diagnóstico diferencialLos cambios radiológicos de la LDM se pueden distinguir de la microangiopatía por envejecimiento o la esclerosis múltiple por su tendencia a ser lesiones simétricas y confluentes en la sustancia blanca1.
Es frecuente que, por las alteraciones en la atención, el lenguaje, el procesamiento de información y las funciones ejecutivas, a algunos pacientes se les puede diagnosticar demencia frontotemporal2; las alteraciones conductuales, asociadas a síntomas sutiles de alteraciones mnésicas y alteraciones motoras, pueden hacer pensar en el diagnóstico de demencia de inicio temprano1.
En los adolescentes, la LDM puede semejarse a una psicosis, debido a que la enfermedad altera el proceso crítico de mielinización, especialmente en la conexiones anatómicas frontotemporales, lo cual desencadena alteraciones en la estructura y la función del sistema nervioso central, lo cual conlleva que se presenten síntomas similares a los de la esquizofrenia2.
Hallazgos imagenológicosEntre los hallazgos en la RM, se incluyen áreas hiperintensas de desmielinización difusa, bilateral y a menudo simétrica en la sustancia blanca periventricular y del cerebelo que pueden confluir con la progresión de la enfermedad, con predominio frontal en las etapas tardías (formas juveniles y en adultos)2,7,12.
En la TC se observan hiperdensidades en sustancia blanca, particularmente en las regiones frontal y parietal. A medida que progresa, se observa atrofia cortical y subcortical, con dilatación ventricular. Las áreas de hipodensidades reflejan pérdida de la mielinización y acumulación del cerebrósido1. Se ha descrito una apariencia «tigroide» o «en piel de leopardo», con franjas de hipointensidad (materia blanca normal) dentro de áreas hiperintensas en T2 de materia blanca anormal (áreas de desmielinización)7.
Hay evidencia de pérdida significativa de volumen de sustancia gris presente desde estadios tempranos de LDM; en presentación adulta, se da una atrofia cortical general más pronunciada de la sustancia tanto gris como blanca y una disminución cortical más prominente en giro cingulado y lóbulos frontales13.
La RM tiene mayor sensibilidad que la TC en la detección de lesiones en la sustancia blanca y es útil para visualizar lesiones de inicio temprano en regiones de la fosa posterior (tallo cerebral o el cerebelo); además, permite ver la gravedad y la extensión de la enfermedad1.
Se ha documentado pérdida significativa de sustancia gris cortical en los estadios tempranos de la enfermedad; en las formas adultas, se presenta mayor atrofia cortical general, más prominente en giro cingulado y lóbulos frontales13.
En la espectroscopia por RM, generalmente muestra una disminución de N-acetil aspartato y mioinositol y, ocasionalmente, aumento de lactato7.
EpidemiologíaLa LDM ocurre con una frecuencia estimada de 1:40.0001,5, aunque se estima que hasta un 16% de la población general puede tener deficiencia de ARS-A1.
Presentación y cursoDebido a que es una enfermedad rara, heterogénea, con presentación clínica diversa y falta de documentación clínica, neurofisiológica y neurorradiológica, se conoce poco acerca de los factores relacionados con la edad de aparición y el curso, por lo cual es difícil predecir la evolución de cada caso particular4.
Las diferentes formas de evolución se relacionan con la mutación existente5. Como toda enfermedad metabólica, la LDM puede producir graves daños en el sistema nervioso central, especialmente en las neuronas que son sensibles a cambios metabólicos, los cuales pueden manifestarse en alteración del desarrollo psicomotor, convulsiones y coma cuando la lesión es grave, y con síntomas sutiles como alteraciones cognitivas y conductuales cuando la lesión es leve2. Existe clasificación clínica de los trastornos motores y de lenguaje, referidos a la gravedad de los síntomas (tabla 1).
Según la edad de aparición, se clasifican en los subtipos infantil tardía, juvenil, juvenil tardía y adulta1,5,6,9.
Subtipo infantil tardíoEl subtipo infantil tardío, descrito por Greenfield14 en 1933, es el de mayor prevalencia. Se presenta alrededor del primer y el segundo año de vida y presenta varias etapas. En la primera, se inicia con alteraciones motoras tales como marcha atáxica o pérdida de la capacidad de caminar, hipotonía y alteración en los reflejos profundos; esta etapa tiene una duración promedio de 16 meses. En la segunda etapa, se profundiza la alteración motora, acompañada de alteraciones mentales, cuya duración es de 3 a 6 meses. Durante la tercera etapa aparece tetraplejía, parálisis bulbar, atrofia del nervio óptico y afección del sistema nervioso central, con una duración de 3 meses a 3 años, seguido de estado vegetativo que puede durar años y generalmente culmina con la muerte, alrededor de los 4–5 años de edad. Las concentraciones de ARS-A son bajas o nulas1,6.
Subtipo juvenilEn la forma juvenil, la edad de aparición es generalmente entre los 4 y los 6 años, se presenta con alteraciones mentales, como labilidad emocional, euforia y cambios del comportamiento, pérdida de las funciones mentales y alteración en el lenguaje que repercuten en el desarrollo intelectual y la actividad escolar. En casos de evolución lenta, predominan los síntomas motores, puede aparecer tetraplejía espástica, síntomas bulbares, ataxia, convulsiones y atrofia del nervio óptico. La duración de la enfermedad puede variar de 3 a 17 años1,6. La actividad enzimática de ARS-A es escasa, pero no tanto como en la forma infantil5.
Subtipo juvenil tardíoEn el subtipo juvenil tardío la enfermedad comienza generalmente entre los 6 y los 16 años, con evolución lenta; se presenta generalmente con problemas de comportamiento, trastornos mentales de evolución lenta y convulsiones o alteraciones motoras. La ARS-A muestra una actividad residual1,5.
Subtipo del adultoEn el subtipo del adulto los síntomas aparecen después de los 16 años. Puede tener dos presentaciones: una de predominio motor (síndrome cerebeloso piramidal) y otro con predominio de síntomas psiquiátricos. Esta forma tardía está fuertemente asociada con mutación de I179S1,2.
En más de la mitad de los pacientes la edad de inicio de la enfermedad se presenta entre los 10 y los 30 años, con síntomas parecidos a la psicosis, enre ellos alucinaciones auditivas, delirios, alteraciones del pensamiento y catatonía2.
La enfermedad progresa con síntomas neurológicos: convulsiones, corea y distonía que se presenta tras el inicio de los síntomas psiquiátricos2.
TratamientoAcualmente no hay tratamiento específico para esta enfermedad, salvo el trasplante de médula ósea en algunos casos seleccionados.
El tratamiento indicado es sintomático y de apoyo; se indican fisioterapia, terapia respiratoria, nutrición, cuidados de enfermería, adaptación de entorno y mantener la red de apoyo y la actividad habitual4. Se ha indicado además el uso de fármacos antiepilépticos y antiespasmódicos4.
La terapia génica se ha mostrado más eficaz que el trasplante tradicional de donante sano para corregir la enfermedad4.
El trasplante de células madre hematopoyéticas (HSCT) es una opción viable, aunque con limitaciones, que puede beneficiar a los fenotipos de inicio tardío en fase temprana, presintomática15.
DiscusiónSe han publicado reportes de caso, series de casos y cohortes14,16–18; el presentado es una LDM del subtipo juvenil tardío que muestra el inicio inespecífico de los síntomas, lo cual hace pensar inicialmente en trastorno psicótico; los exámenes iniciales fueron negativos, incluso las imágenes diagnósticas; el control sintomático inadecuado y el curso deteriorante llevaron a necesidad de cuidados permanentes en institución; posteriormente se evidenció la aparición de signos de tipo motor que hacían sospechar enfermedad neurodegenerativa y se realizó el diagnóstico con el concurso de psiquiatría, neurología y genética. El cuadro inicial, dados la edad y los síntomas, hizo pensar en el inicio de una enfermedad psicótica del tipo esquizofrenia, aunque su progresión y la falta de respuesta terapéutica replantearon el diagnóstico.
Si bien se han comunicado casos de LDM en familiares18, la mayoría de los casos, como el presentado aquí, corresponden a solo una persona afectada en el genograma. Los hallazgos inicialmente negativos en la imagenología diagnóstica y posteriormente la presencia de lesiones en la RM evidencian la progresión clínica.
Los psiquiatras deben ser conscientes de su papel en diagnosticar precozmente la LDM, dado que los síntomas iniciales pueden llevar a la consulta por psiquiatría; si bien su prevalencia es baja, la aparición puede ser similar a la de la esquizofrenia y otros trastornos psicóticos. Un seguimiento adecuado supone la posibilidad de un diagnóstico y manejo más tempranos.
Aunque no hay evidencia de tratamiento adecuado de la condición y se supone que el tratamiento sintomático y las medidas de apoyo sean clave, es necesario evitar al máximo los efectos secundarios de la medicación.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
