


La hipercolesterolemia familiar es una enfermedad genética que se caracteriza por niveles muy elevados de colesterol y lipoproteínas de baja densidad en suero, xantomas tendinosos y aterosclerosis prematura. La forma heterocigota es la más común; alcanza una prevalencia de aproximadamente 1 de cada 300 a 500 personas en el mundo, en tanto que la homocigota, autosómica dominante, es la forma más rara, con una prevalencia de 1 en 1 millón de personas. Esta se caracteriza por hipercolesterolemia severa, que conlleva enfermedad cardiovascular prematura y a menudo no responde al tratamiento tradicional por la falta de receptores para c-LDL funcionales. Los niveles de c-LDL pueden superar seis a diez veces los valores normales, en cuyo caso el trasplante de hígado se ha convertido en el tratamiento de elección para los pacientes que no responden a tratamientos farmacológicos de rutina. Se presentan dos casos con hipercolesterolemia familiar homocigota en jóvenes de 14 y 15 años, con antecedente de trasplante de hígado y enfermedad coronaria severa en vasos principales (descendente anterior y coronaria derecha) a quienes se les hizo implante exitoso de stent liberador de medicamento.
Familial hypercholesterolemia is a genetic disorder characterised by very high cholesterol and low-density lipoproteins serum levels, tendon xanthomas and premature atherosclerosis. Heterozygous form is the most common, with a prevalence of approximately 1 out of 300 to 500 people worldwide, whereas the homozygous, autosomal dominant, is the rarest form, with a prevalence of 1 out of 1 million people. It is characterised by severe hypercholesterolemia leading to premature cardiovascular disease, and it often does not respond to traditional therapy due to the lack of receptors for functional LDL-c. LDL-c levels can exceed between six and ten times the normal values, in which case liver transplantation has become the treatment of choice for patients who do not respond to routine pharmacological therapies. This study presents two cases of homozygous familial hypercholesterolemia in young patients aged 14 and 15, with prior liver transplantation and severe coronary disease in major vessels (anterior descending artery and right coronary artery) who underwent successful implant of a drug-eluting stent.
Los trastornos del metabolismo de las lipoproteínas, así como las dietas ricas en grasas, la obesidad y la inactividad física, han dado lugar a una epidemia mundial de enfermedad aterosclerótica. La interacción de los trastornos genéticos y adquiridos de las lipoproteínas con estos factores ambientales adversos, predispone a aterosclerosis prematura. En los Estados Unidos, la mortalidad por enfermedad coronaria, principalmente en personas de mediana edad, ha disminuido en un 31% en la última década; sin embargo, la enfermedad cardiovascular aterosclerótica sigue siendo la causa más común de muerte tanto en hombres como en mujeres. La tasa general de muertes atribuibles a enfermedades cardiovasculares es de 235,5 por 100.000 habitantes y la enfermedad coronaria sola causó 1 de cada 6 muertes en los Estados Unidos en el 20101.
La hipercolesterolemia familiar (HF) es una enfermedad genética que se caracteriza por niveles muy elevados de colesterol y lipoproteínas de baja densidad en suero, xantomas tendinosos y aterosclerosis prematura2. Más del 85% de los casos de HF se deben a más de 1.600 mutaciones heredadas en el gen del receptor LDL (R-LDL). Dicha mutación conduce a la captación defectuosa del c-LDL de la sangre3–5.
La HF heterocigota es la forma más común de la enfermedad (prevalencia de aproximadamente 1 de cada 300 a 500 personas en todo el mundo y tan alta como 1 de cada 100 personas en algunas poblaciones)3–6, mientras que la HF homocigota, autosómica dominante, es la forma más rara (prevalencia de 1 en 1 millón de personas). Algunas poblaciones, como los franco-canadienses, judíos Ashkenazi, libaneses y holandeses de origen africano, están en riesgo más alto para HF debido a un aumento de la prevalencia de mutaciones asociadas a HF heterocigota7–10.
La variable homocigota se caracteriza por hipercolesterolemia severa que conduce a enfermedad cardiovascular prematura. Los individuos tienen mayor riesgo de eventos cardiacos como infarto de miocardio y muerte por enfermedad coronaria prematura, especialmente aquellos con formas severas que no han sido tratados11,12. Constituye una forma grave y agresiva de la enfermedad, que a menudo no responde al tratamiento tradicional dada la falta de receptores para c-LDL funcionales12,13. En general, la edad promedio en el momento del diagnóstico de las manifestaciones cardiovasculares es 20 años12. La elevación de los niveles de c-LDL refleja la severidad de la mutación genética. En pacientes con HF heterocigota típicamente se presentan niveles al doble o triple en comparación con individuos sanos (aproximadamente 200-400mg/dl), mientras que los pacientes con HF homocigota tienen niveles de c-LDL que pueden superar seis a diez veces los valores normales (> 600mg/dl)13.
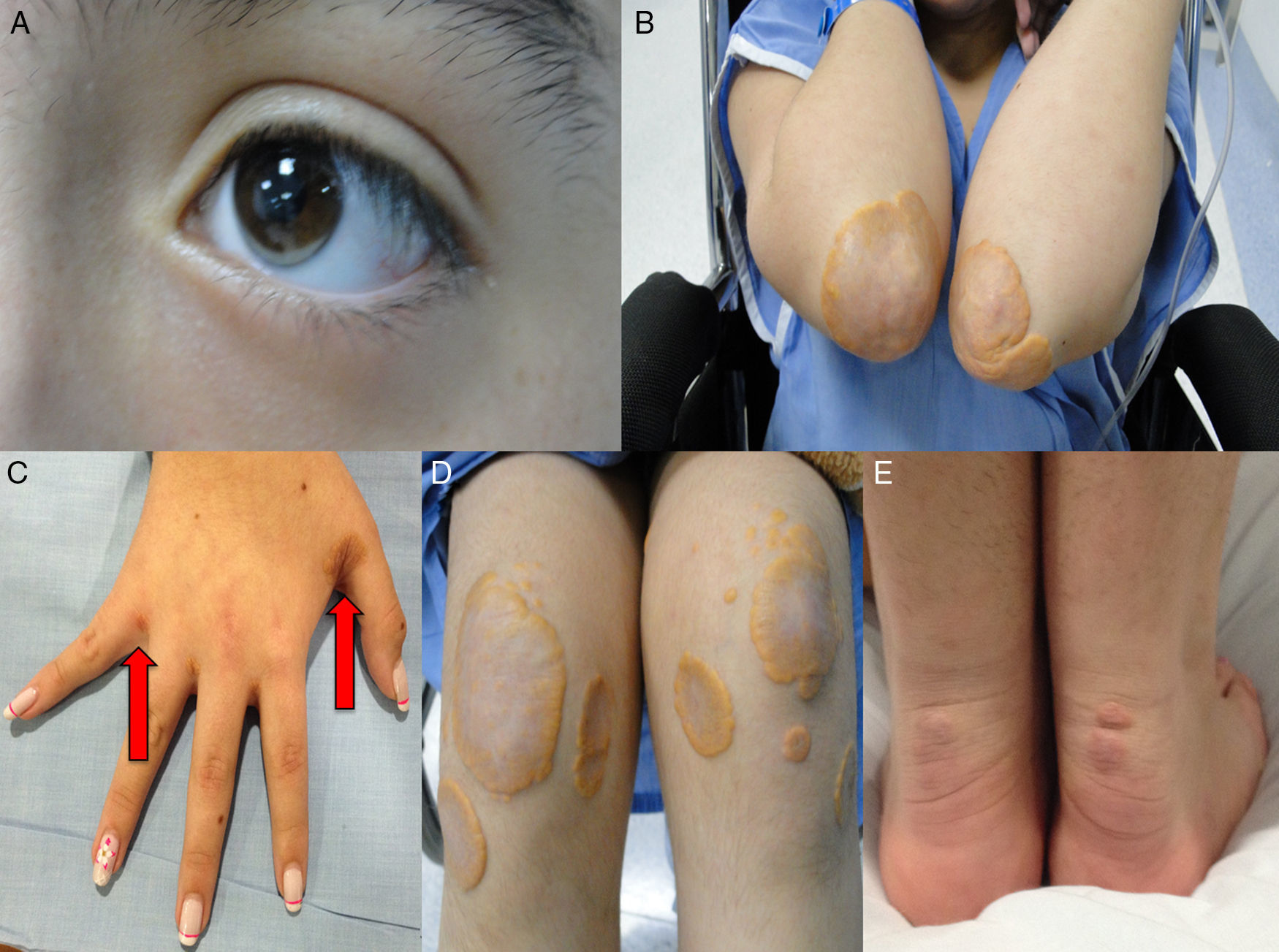
Así mismo, la forma homocigota se asocia con enfermedad coronaria y muerte prematura; de hecho existen varios informes en menores de 17 años de edad que desarrollaron estenosis coronaria severa2,14 y estenosis aórtica supravalvular15, aumento del grosor de la íntima-media de la carótida y arteria femoral16 y xantomas tendinosos causados por el depósito de colesterol en los tendones y la piel, que se observan principalmente en codos, rodillas, tendón de Aquiles, y dorso de manos y pies. Son altamente sugestivos de HF homocigota y son un criterio básico en el diagnóstico clínico de esta enfermedad. En pacientes con HF existen xantomas hasta en el 50% de los casos17. El colesterol en exceso también se puede depositar en la córnea, dando lugar al arco corneal. Para conocer más a fondo sobre esta patología lo invitamos a consultar el artículo especial de revisión sobre HF publicado por Merchán et al.18.
Este artículo obedece a las características no comunes en el tratamiento de pacientes con HF homocigota como lo son la enfermedad aterosclerótica progresiva en adolescentes luego de trasplante hepático exitoso y el control de su perfil lipídico, que obligan a un manejo interdisciplinario complejo, así como a un seguimiento estricto.
Pacientes y métodosSe describe la experiencia de dos pacientes jóvenes que acudieron al Servicio de Cardiología Intervencionista de la institución, en septiembre y octubre de 2013, con características clínicas de HF homocigótica, trasplante hepático y enfermedad coronaria severa, y fueron sometidos a revascularización miocárdica percutánea con implante de stent liberadores de fármaco.
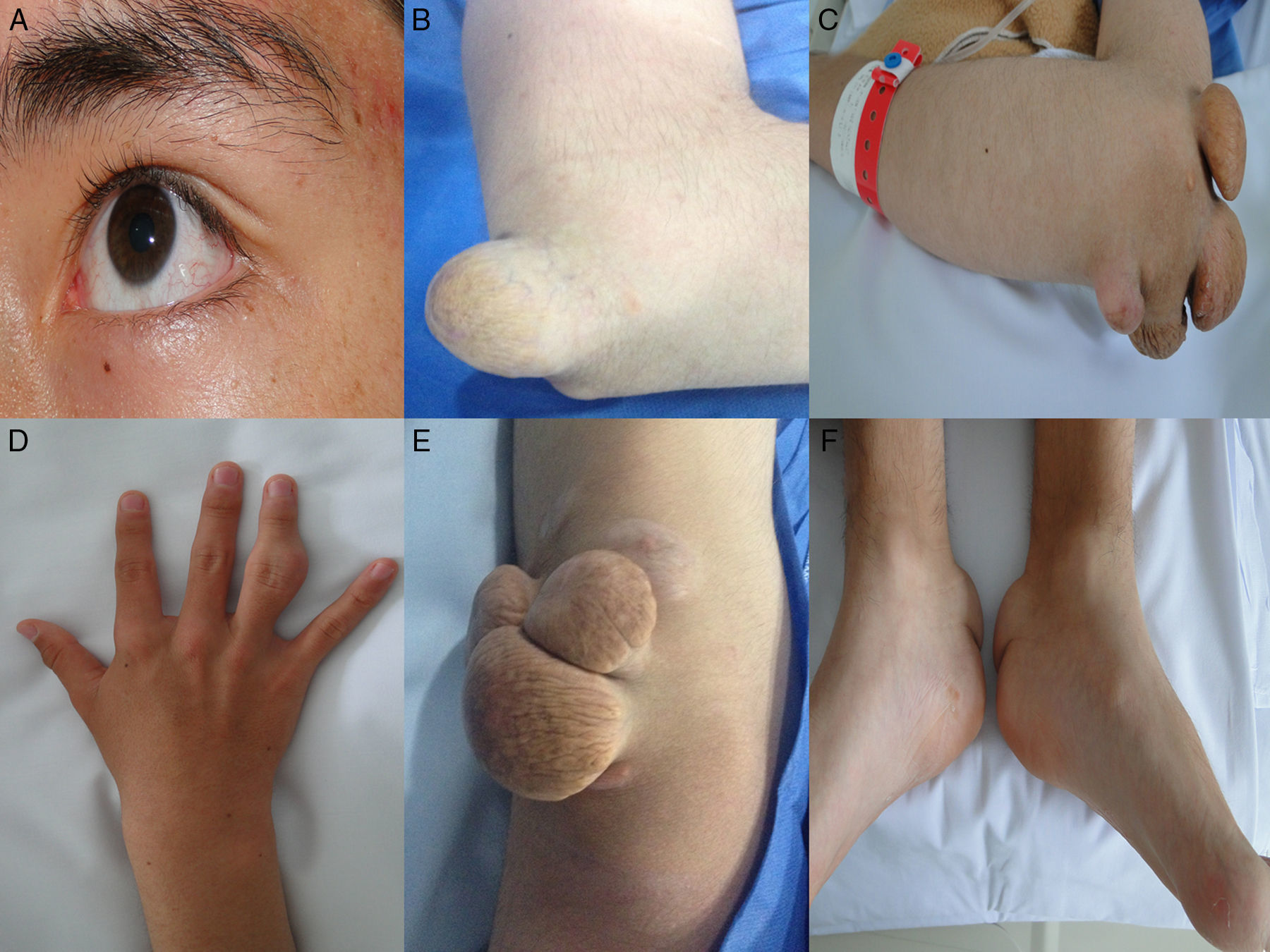
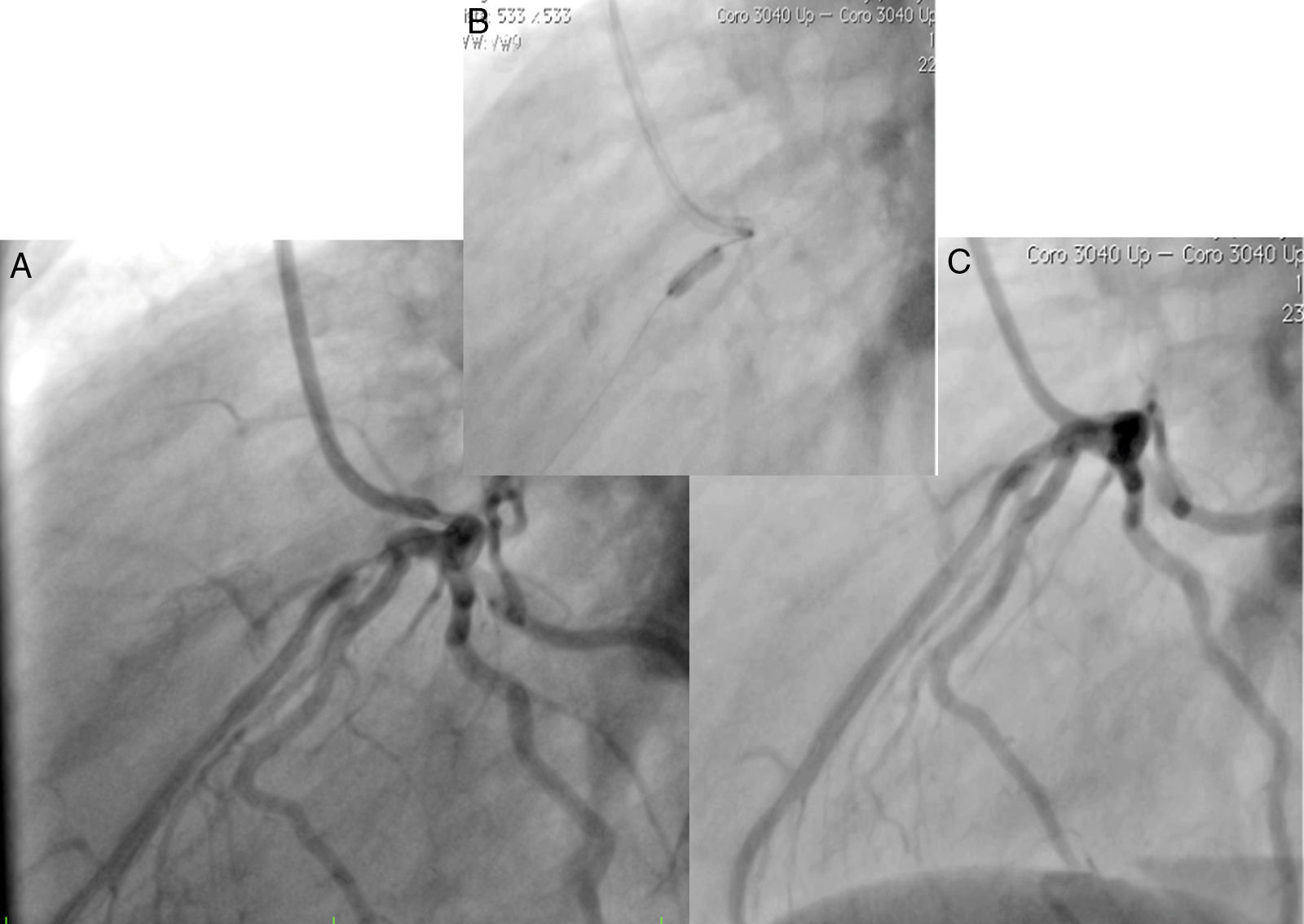
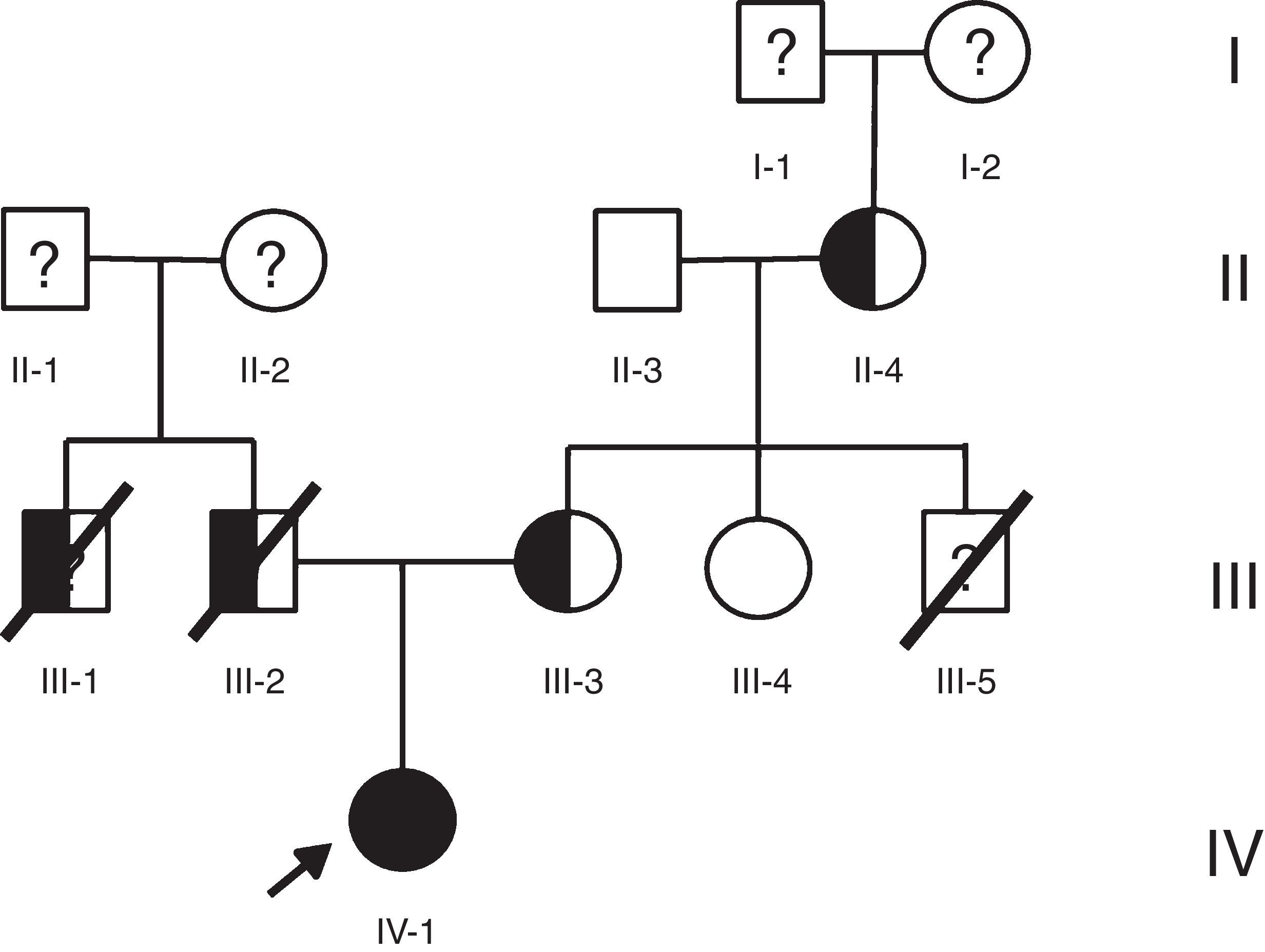
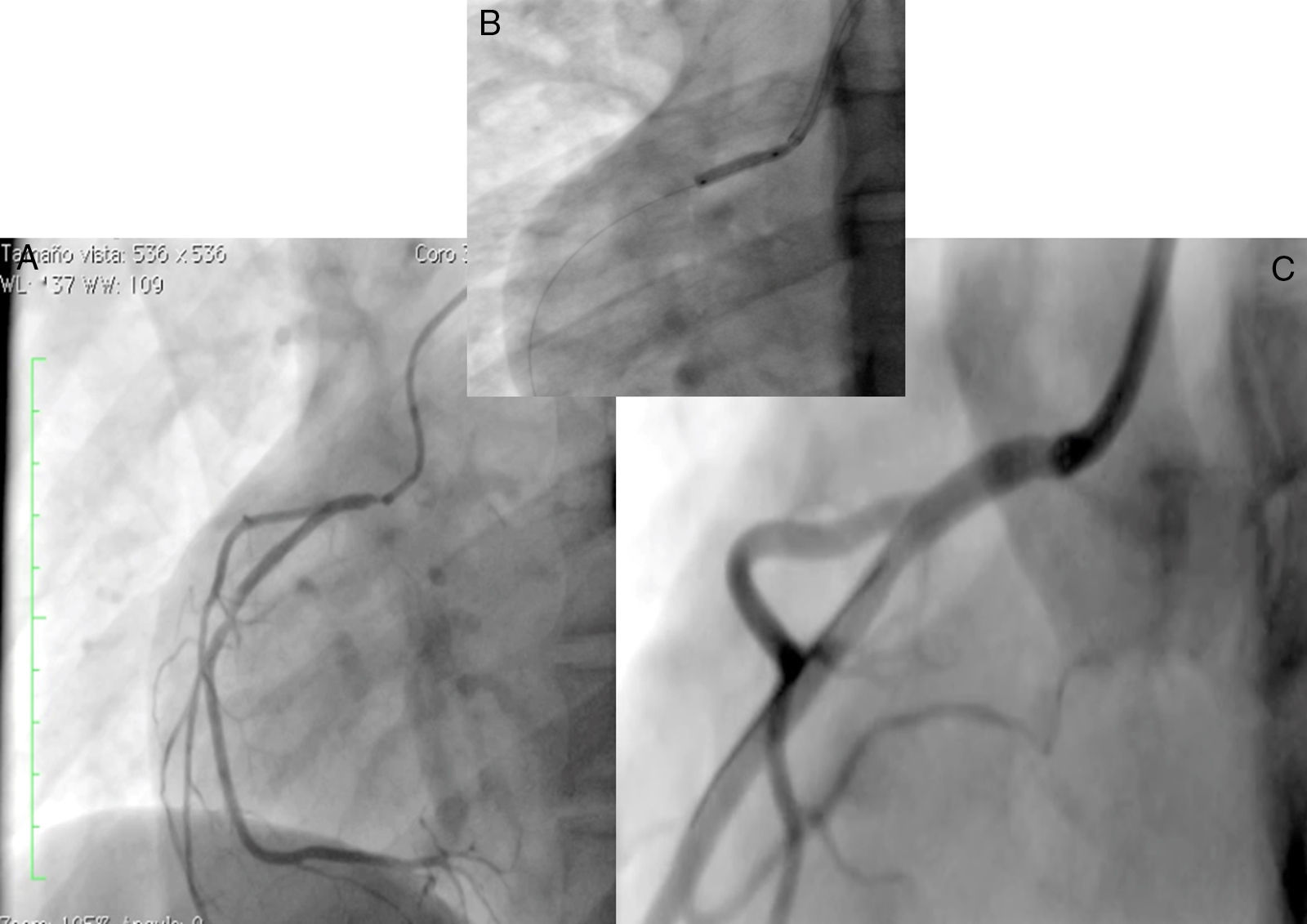
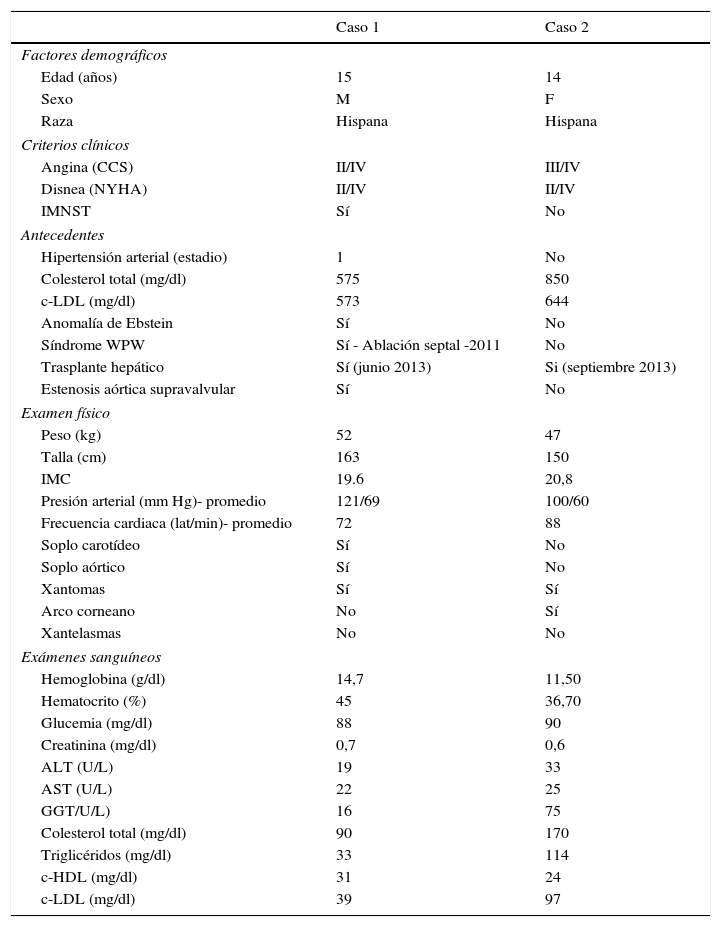
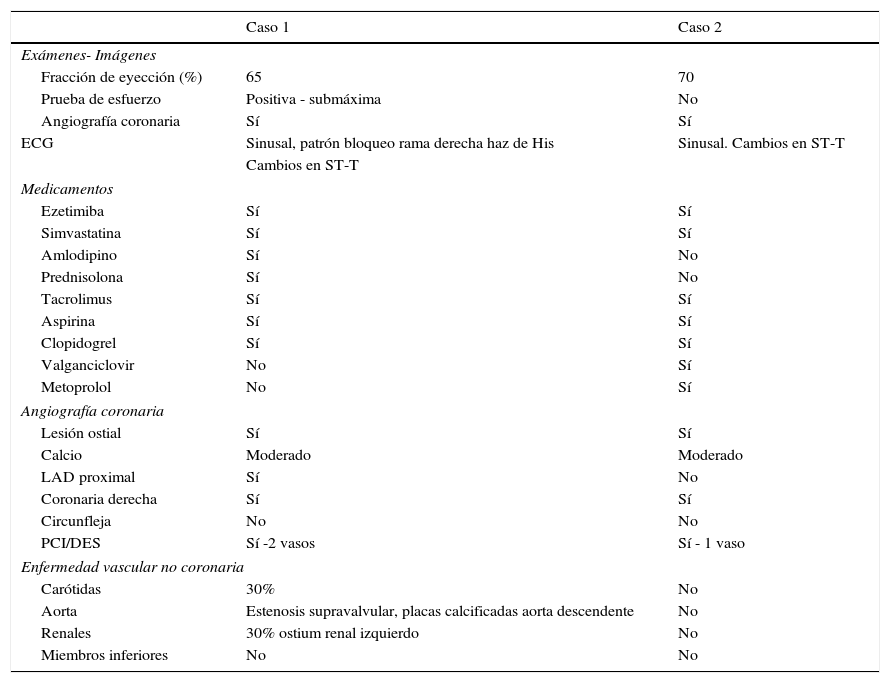
Caso 1Hombre de 15 años de edad (tablas 1 y 2), con cuadro de angina de pecho de dos semanas de evolución, deterioro de clase funcional, infarto sin elevación del ST, troponina (+), antecedente de HF homocigota (fig. 1), xantomas cutáneos tuberosos (fig. 2), hiperlipidemia severa (c-LDL hasta de 850mg/dl), hipertensión arterial estadio 1; anomalía de Ebstein y síndrome de Wolff-Parkinson-White ablactado en septiembre de 2011, sometido a trasplante de hígado en junio-2013, y prueba de esfuerzo submáxima positiva para isquemia. Se hallaba en tratamiento farmacológico con ezetimiba 10mg/día, simvastatina 20mg/día, amlodipino 5mg/día, prednisolona 2,5mg/día y tacrolimus 3mg/día. La angiografía coronaria reveló enfermedad coronaria severa en arteria descendente anterior proximal y coronaria derecha ostial. Se realizó angioplastia coronaria con implante de stent liberador de medicamento Resolute Integrity™(Medtronic Vascular, Santa Rosa, CA) 3,0×12mm y 2,25×15mm respectivamente, post-dilatación e impactación con balón no complaciente a 16 atms (figs. 3 y 4). En seguimiento clínico a tres meses se le encontró asintomático y adscrito al programa de rehabilitación cardiaca.
Manifestaciones demográficas, clínicas y paraclínicas de los dos casos con hipercolesterolemia familiar homocigota
| Caso 1 | Caso 2 | |
|---|---|---|
| Factores demográficos | ||
| Edad (años) | 15 | 14 |
| Sexo | M | F |
| Raza | Hispana | Hispana |
| Criterios clínicos | ||
| Angina (CCS) | II/IV | III/IV |
| Disnea (NYHA) | II/IV | II/IV |
| IMNST | Sí | No |
| Antecedentes | ||
| Hipertensión arterial (estadio) | 1 | No |
| Colesterol total (mg/dl) | 575 | 850 |
| c-LDL (mg/dl) | 573 | 644 |
| Anomalía de Ebstein | Sí | No |
| Síndrome WPW | Sí - Ablación septal -2011 | No |
| Trasplante hepático | Sí (junio 2013) | Si (septiembre 2013) |
| Estenosis aórtica supravalvular | Sí | No |
| Examen físico | ||
| Peso (kg) | 52 | 47 |
| Talla (cm) | 163 | 150 |
| IMC | 19.6 | 20,8 |
| Presión arterial (mm Hg)- promedio | 121/69 | 100/60 |
| Frecuencia cardiaca (lat/min)- promedio | 72 | 88 |
| Soplo carotídeo | Sí | No |
| Soplo aórtico | Sí | No |
| Xantomas | Sí | Sí |
| Arco corneano | No | Sí |
| Xantelasmas | No | No |
| Exámenes sanguíneos | ||
| Hemoglobina (g/dl) | 14,7 | 11,50 |
| Hematocrito (%) | 45 | 36,70 |
| Glucemia (mg/dl) | 88 | 90 |
| Creatinina (mg/dl) | 0,7 | 0,6 |
| ALT (U/L) | 19 | 33 |
| AST (U/L) | 22 | 25 |
| GGT/U/L) | 16 | 75 |
| Colesterol total (mg/dl) | 90 | 170 |
| Triglicéridos (mg/dl) | 33 | 114 |
| c-HDL (mg/dl) | 31 | 24 |
| c-LDL (mg/dl) | 39 | 97 |
Manifestaciones demográficas, clínicas y paraclínicas de los dos casos con hipercolesterolemia familiar homocigota
| Caso 1 | Caso 2 | |
|---|---|---|
| Exámenes- Imágenes | ||
| Fracción de eyección (%) | 65 | 70 |
| Prueba de esfuerzo | Positiva - submáxima | No |
| Angiografía coronaria | Sí | Sí |
| ECG | Sinusal, patrón bloqueo rama derecha haz de His | Sinusal. Cambios en ST-T |
| Cambios en ST-T | ||
| Medicamentos | ||
| Ezetimiba | Sí | Sí |
| Simvastatina | Sí | Sí |
| Amlodipino | Sí | No |
| Prednisolona | Sí | No |
| Tacrolimus | Sí | Sí |
| Aspirina | Sí | Sí |
| Clopidogrel | Sí | Sí |
| Valganciclovir | No | Sí |
| Metoprolol | No | Sí |
| Angiografía coronaria | ||
| Lesión ostial | Sí | Sí |
| Calcio | Moderado | Moderado |
| LAD proximal | Sí | No |
| Coronaria derecha | Sí | Sí |
| Circunfleja | No | No |
| PCI/DES | Sí -2 vasos | Sí - 1 vaso |
| Enfermedad vascular no coronaria | ||
| Carótidas | 30% | No |
| Aorta | Estenosis supravalvular, placas calcificadas aorta descendente | No |
| Renales | 30% ostium renal izquierdo | No |
| Miembros inferiores | No | No |

, Xantomas tuberosos en codos (A y B), manos (C), rodillas (D) y tendón de Aquiles (F).")
, implante de stent liberador de medicamento (B) y resultado final (C).")
, implante de stent liberador de medicamento (B) y resultado final (C).")
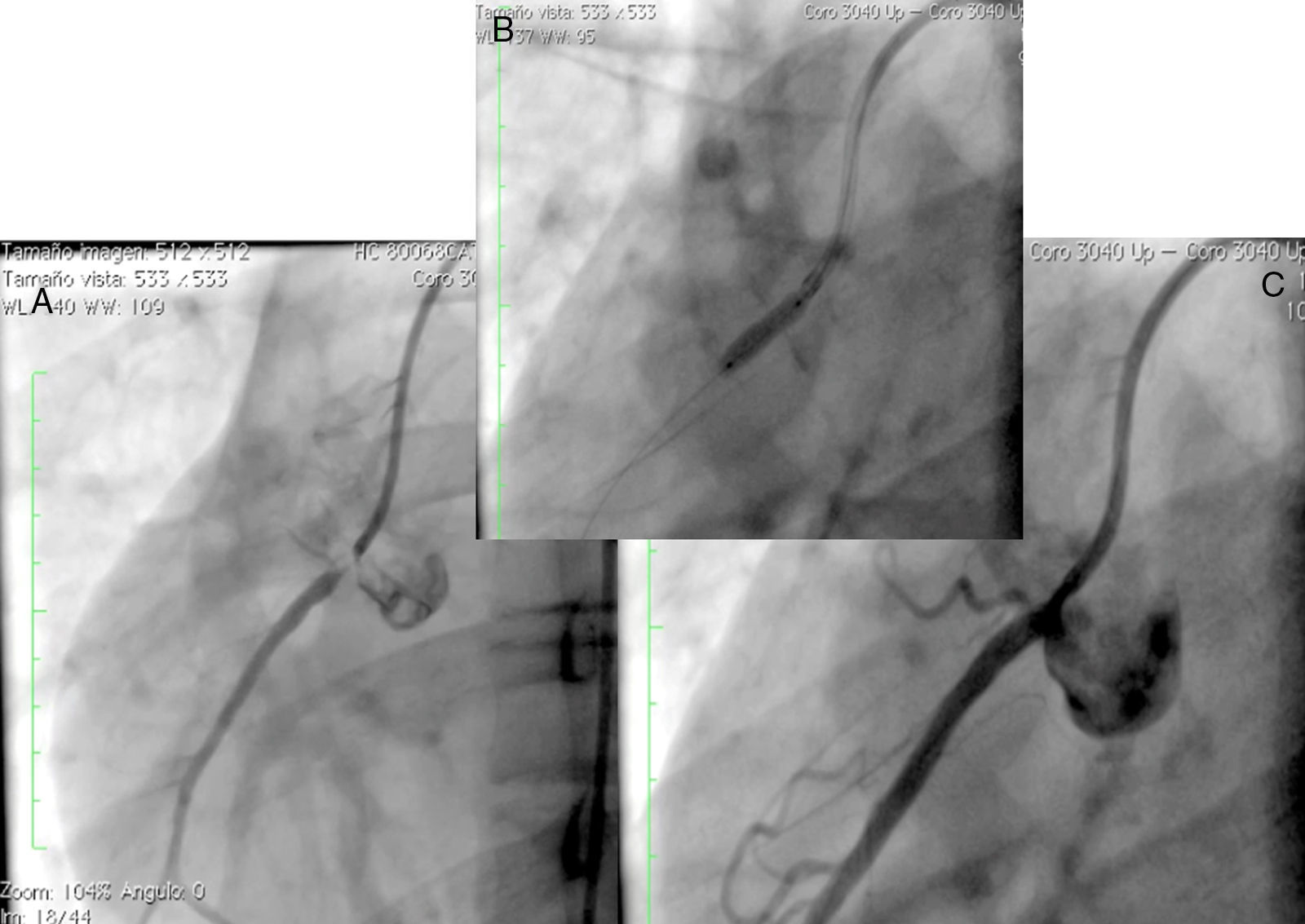
Mujer de 14 años (tablas 1 y 2), con angina de pecho inestable y disnea de inicio reciente y antecedente de HF homocigota (fig. 5). El análisis de historia familiar reveló una historia extensa de HF heterocigota y eventos cardiovasculares en las familias materna y paterna. Se evidenciaron xantomas cutáneos planos y arco corneano (fig. 6), hiperlipidemia severa (c-LDL hasta 644mg/dl) y trasplante de hígado en septiembre de 2013. El análisis genético por secuenciación de los genes LDLR y ApoB identificó una mutación homocigótica o hemicigótica en un intrón de LDLR y una mutación homo- o hemicigótica en un exón de LDLR. Se hallaba en tratamiento con ezetimiba 10mg/día, simvastatina 20mg/día, valganciclovir 1 tab/día y tacrolimus 3mg/día. La angiografía coronaria reveló enfermedad coronaria severa en la coronaria derecha ostial. Se hizo angioplastia coronaria con implante de stent liberador de medicamento Resolute Integrity™ (Medtronic Vascular, Santa Rosa, CA) 2,5×15mm y postdilatación e impactación con balón no complaciente a 16 atms (fig. 7). En el seguimiento clínico a 3 meses se le encontró asintomática y adscrita al programa de rehabilitación cardiaca.

, Xantomas planos en codos (A), manos (C), rodillas (D) y tendón de Aquiles (E).")
, implante de stent liberador de medicamento (B) y resultado final (C).")
Como se expresó antes, la HF es un trastorno genético autosómico dominante, monogénico, causado por mutaciones en el gen del R-LDL. En este artículo se exponen dos casos con HF homocigota en jóvenes de 14 y 15 años y enfermedad coronaria severa en vasos principales (descendente anterior y coronaria derecha) sometidos a implante exitoso de stent liberador de medicamento. Ambos tenían como característica particular, la edad, las manifestaciones cardiovasculares (angina de pecho) y encontrarse en un periodo postrasplante de hígado (3 y 2 meses respectivamente) sin evidencia de rechazo, además de perfil de lípidos en niveles normales.
La literatura describe que en pacientes con HF homocigota, la edad media en el momento del diagnóstico de las enfermedades cardiovasculares es 20 años11,19. Sin embargo, varias publicaciones correspondientes a la década de los 80 describen pacientes homocigotos menores de 17 años de edad que desarrollaron enfermedad coronaria severa y estenosis aórtica supravalvular, fecha para cual la terapia de revascularización coronaria percutánea con stents estaba en sus inicios2,15,20,21.
Tellez et al.22 describieron en 2010 la respuesta vascular que ocurre después del implante de un stent metálico en un modelo porcino con HF, demostrando un comportamiento en la formación neointimal más agresivo que el que ocurre en animales domésticos, lo cual plantea la necesidad de considerar el uso de stents liberadores de medicamento en condiciones de HF. Recientemente, Shankarappa et al14 reportaron los casos de 5 pacientes, 4 hombres y 1 mujer, tratados en el Instituto Sri Jayadeva de Cardiología, en Bangalore, India. Todos presentaron síntomas coronarios, xantomas tendinosos y enfermedad coronaria severa, además de cifras de colesterol total mayores a 435mg/dl y c-LDL mayores a 392mg/dl. Tres de ellos requirieron cirugía de revascularización arterial y los otros dos, angioplastia con implante exitoso de stents. En un hombre de 24 años se implantó stent liberador de medicamento en descendente anterior y stent metálico en circunfleja, y en una mujer de 17 años stent liberador de medicamento en tronco principal izquierdo.
La intervención coronaria percutánea (ICP) se utiliza comúnmente en pacientes adultos con enfermedad coronaria severa. En aquellos con angina inestable o infarto sin elevación del ST, se indica estrategia invasiva precoz (clase I, nivel de evidencia B)23. De otra parte, la información de ICP en niños y adolescentes es muy limitada. La mayoría de los casos descritos en menores de 18 años, han tenido indicación no aterosclerótica, como vasculopatía coronaria del trasplante cardiaco24–27, disección coronaria24 y compresión u oclusión coronaria como secuela de cirugía de corazón previa24,28,29.
Las Guías publicadas por la US National Lipid Association (NLA) y NICE en el Reino Unido, recomiendan una reducción en la concentración de c-LDL de más del 50% respecto a los niveles previos al tratamiento en pacientes con HF30–33. Las Guías de Canadá y Europa recomiendan la reducción de los niveles de c-LDL a menos de 116mg/dl en pacientes con riesgo moderado para enfermedad cardiovascular; menos de 97mg/dl en pacientes de alto riesgo, y menos de 70mg/dl en pacientes de muy alto riesgo.
Hoy día se dispone de diferentes opciones para el tratamiento de los pacientes afectados por HF, entre ellas, dieta, terapia farmacológica, aféresis de lípidos y algunas técnicas quirúrgicas como la cirugía de derivación portocava que limita la absorción de colesterol y promueve la pérdida de ácidos biliares, y el trasplante de hígado; para disponer de Rs-LDL funcionales, este último constituye una alternativa para los casos más graves34.
Pocos años después que Brown y Goldstein ganaran el premio Nobel de Medicina en 1985 al describir que la HF se debía a un defecto intrínseco del hepatocito en la deficiencia de receptores de c-LDL como explicación de las cifras extremadamente altas de colesterol sérico21, Starzl et al.35 reportaron el primer trasplante hepático ortotópico realizado fuera de las indicaciones habituales de una insuficiencia hepática, como tratamiento de la hiperlipidemia en el contexto de HF homocigótica. Fue la primera vez que un hígado anatómicamente normal se extirpaba quirúrgicamente en una paciente de seis años de edad, para el tratamiento presuntivo de la condición hipercolesterolémica.
Como bien se sabe, la mayoría de los R-LDL se encuentran en el hígado, por ello el trasplante de hígado se ha convertido en el tratamiento de elección para los pacientes afectados que no responden a tratamientos farmacológicos de rutina36,37. El hígado trasplantado conserva las cualidades específicas del donante, de modo que el trasplante puede ser fuente abundante de receptores de LDL funcionales, y conducir a la cura de la hipercolesterolemia34. Gracias a los avances en la experiencia por parte de los grupos de trasplante, la inmunosupresión a largo plazo, mejoras en las técnicas quirúrgicas y los métodos inmunológicos, se han alcanzado resultados favorables después del trasplante tanto en población adulta como pediátrica35,38. Los casos expuestos tenían al momento de su intervención coronaria, trasplante hepático exitoso y cifras de colesterol normales.
Conclusiones y recomendacionesLa HF es un trastorno genético autosómico dominante, asociado con niveles elevados de c-LDL, que puede conducir a enfermedad cardiovascular prematura. El diagnóstico precoz es crucial para prevenir la morbilidad y la mortalidad. Este generalmente se hace con base en las características clínicas, los antecedentes familiares y los niveles séricos de colesterol. Las guías actuales destacan la importancia de reducir los niveles de c-LDL en pacientes con HF. En este documento se informaron los casos de dos pacientes de 14 y 15 años con manifestaciones clínicas de HF homocigota severa tratada con fármacos y trasplante hepático, que además presentaron síndrome coronario agudo sin elevación del ST y enfermedad coronaria severa tratada de manera exitosa con implante de stents liberadores de medicamento.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interésLos autores declaran no tener conflictos de interés.
Siguiendo la política de publicación de la revista, este artículo, en el que ha participado como coautor un miembro del Comité editorial, ha superado un estricto proceso de revisión por pares doble ciego y los autores no han tenido ninguna injerencia en la aprobación para su publicación.