




la hipercolesterolemia familiar representa un factor de riesgo sustancial para padecer enfermedad coronaria prematura, arterial periférica y valvular. Se han descrito dos formas según su alteración genética y cigocidad, así como tres mutaciones genéticas asociadas. Pese a que el tratamiento con estatinas se considera la primera línea, algunos pacientes no alcanzan metas, de modo que se han utilizado los inhibidores del PCSK9 como nueva estrategia.
Métodos y materialesse expone el caso de una paciente de 42 años con hipercolesterolemia familiar heterocigota tratada con inhibidores del PCSK9. Se describen los criterios y estudios genéticos utilizados para realizar el diagnóstico, la cronología de tratamientos que recibió y los exámenes de laboratorio anteriores y posteriores al inicio del evolocumab. Adicionalmente se hace una revisión de tema acerca de la hipercolesterolemia familiar y su tratamiento con inhibidores del PCSK9.
Conclusionesla hipercolesterolemia familiar es una enfermedad que ocasiona graves consecuencias cardiovasculares. Los inhibidores del PCSK9 se han convertido en una alternativa prometedora para aquellos que no responden a las terapias convencionales. Se requieren estudios que corroboren o contradigan los beneficios y eventos adversos encontrados hasta el momento en que los pacientes se someten a estas nuevas terapias para así ofrecer un tratamiento ideal y oportuno.
Familial hypercholesterolaemia is a substantial risk factor for suffering premature coronary, peripheral arterial, and valular disease. There are two forms described, depending on their genetics and zygosity, as well as three associated genetic mutations. Although treatment with statins is considered first line, some patients do not reach targets, as such that that PCSK9 inhibitors have been used as a new strategy.
Materials and methodA case is presented of a 42 year-old patient with heterozygous familial hypercholesterolaemia treated with PCSK9 inhibitors. The criteria and genetic studies used to make a diagnosis are described, as well as the chronology of the treatments that have been received and the laboratory results before and after starting with evolocumab. A review has also been made of the subject of familial hypercholesterolaemia and its treatment with PCSK9 inhibitors.
ConclusionsFamilial hypercholesterolaemia is a diseases that may have serious cardiovascular consequences. PCSK9 inhibitors have become a promising alternative for those who do not respond to conventional therapies. Studies are required that can corroborate or contradict the benefits and adverse effects found up until now in patients subjected to these new therapies in order to offer an ideal and appropriate treatment.
La hipercolesterolemia familiar es un trastorno genético autosómico dominante que se caracteriza por un aumento importante de los niveles de colesterol de baja densidad (c-LDL, su sigla en inglés), y a su vez genera un incremento significativo en el riesgo cardiovascular, acompañado de sus patologías y complicaciones consecuentes.
Dentro de la hipercolesterolemia familiar se enmarcan dos tipos: la homocigota y la heterocigota. En ambos casos, la enfermedad coronaria aparece antes que en el resto de la población.
Se ha calculado que entre 14 y 34 millones de personas en el mundo padecen hipercolesterolemia familiar, sin embargo, en menos del 1% de los afectados se logra establecer el diagnóstico1, lo cual implica que muchos pacientes no reciban las medidas terapéuticas adecuadas para tratar una enfermedad que suele ser muy agresiva para adultos jóvenes y en edad media.
Se sabe que existen tres mutaciones genéticas asociadas con la hipercolesterolemia familiar: la del receptor de LDL (R-LDL), la de la apolipoproteína B (ApoB) y la de la pro-proteína convertasa subtilina/kenina 9 (PCSK9). Gracias a que se han reconocido dichas mutaciones, se han podido desarrollar terapias farmacológicas que buscan un impacto en aquellos pacientes que no responden a los medicamentos de uso habitual para hipercolesterolemias sin origen genético. Dentro de esta nueva gama de medicamentos están los inhibidores de PCSK9, anticuerpos monoclonales que buscan influenciar la reducción de niveles de c-LDL.
A continuación se presenta el caso de una paciente joven con hipercolesterolemia familiar heterocigota, diagnosticada mediante estudio genético, con importantes compromisos cardiovasculares y no respuesta a tratamientos farmacológicos convencionales, razón por la que se decidió iniciar terapia con inhibidores de PCSK9.
CasoPaciente femenina, de 42 años, raza mestiza, madre de dos hijas, con antecedente de dislipidemia diagnosticada a los 32 años y en tratamiento con estatinas, sin historia de consumo de sustancias psicoactivas, alcohol, tabaquismo, sobrepeso u obesidad. Como antecedentes familiares relató enfermedad coronaria y dislipidemia en madre, hermana y tíos maternos (entre ellas dos mujeres); estos últimos murieron de infarto agudo de miocardio antes de los 50 años. Su madre tuvo el primer infarto agudo de miocardio a los 55 años y es diabética insulino-requiriente. Ambas hijas, adolescentes, tienen dislipidemia documentada.
Fue valorada por primera vez por el grupo CES Cardiología en 2014, fecha en la que consultó para continuar manejo de su dislipidemia, dolor en el miembro superior derecho y dolor precordial de varios meses de evolución.
Debido al cuadro clínico y sus antecedentes personales, se hicieron varios estudios e intervenciones. Se llevó a arteriografía selectiva de miembros superiores en la que se documentó síndrome del opérculo torácico bilateral con compromiso severo de la extremidad superior derecha. También se realizó arteriografía coronaria en la que se evidenció compromiso severo del ostium de la primera rama diagonal (ramus intermedio), y así mismo se hizo angioplastia con doble balón e implantación de un stent medicado en el tercio medio de la arteria coronaria descendente anterior y primera rama diagonal. La arteria coronaria derecha presentaba enfermedad aterosclerótica difusa sin repercusión hemodinámica.
Debido a estos hallazgos y al cuadro clínico sugestivo de hipercolesterolemia familiar, se aplicaron los criterios de la escala holandesa “Dutch lipid clinic network” para hipercolesterolemia familiar (tabla 1), la cual arrojó un puntaje de 10 y apoyó el diagnóstico definitivo con base en los siguientes criterios: familiar en primer grado con enfermedad coronaria, familiar en primer grado con dislipidemia, menores de 18 años con anormalidad en el c-LDL, enfermedad coronaria prematura, enfermedad vascular periférica y niveles actuales de c-LDL entre 191-250mg/dl. Se estableció que el riesgo cardiovascular global por la escala de Framingham era del 39%.
Criterios de “Dutch lipid clinic network” para hipercolesterolemia familiar, aplicados al caso
| Criterios que cumple la paciente | Puntaje |
|---|---|
| Infarto agudo de miocardio en madre de la paciente a los 55 años por enfermedad coronaria. Requirió manejo con bypass coronario. | 1 |
| Hija de la paciente con hipercolesterolemia: Hija #1: 10 años, c-LDL* 141mg/dl. | 1 |
| Menor de 18 años con c- LDL en percentil superior para edad y género (hija #1). | 1 |
| Paciente con enfermedad coronaria prematura (mujer menor de 60 años): implante de 2 stents medicados | 2 |
| c-LDL entre 191 y 250mg/dl | 3 |
| Total | 8 |
Cabe anotar que el diagnóstico confirmatorio de hipercolesterolemia familiar debe realizarse con un estudio genético, prueba de oro para estos casos. En concordancia, este se solicitó al grupo GENOMACES, quienes reportaron tres mutaciones asociadas a dislipidemia, ubicadas en los genes APOA5, APOE y SCARB1. Adicionalmente, la paciente tiene otras mutaciones genéticas; pese a estos cambios no se ha reportado asociación clínica en las bases de datos genómicas, si bien otros cambios genéticos dentro del mismo gen se han relacionado directamente con las dislipidemias. Dichas mutaciones se encuentran en los genes: INSRR (1 mutación, 1q23.1), APOB (1 mutación, 2p24.1), GCKR (2p23.3), ABCA12 (2q35), CELSR3 (3p21.31), MTTP (4q23), ABCA13 (7p12.3), LDLRAD3 (11p13), CELA1 (12q13.13), LMF1 (16p13.3), APOBR (16p11.2), ABCA10 (17q24.3), INSR (19p13.2) y CELSR1 (22q13.31).
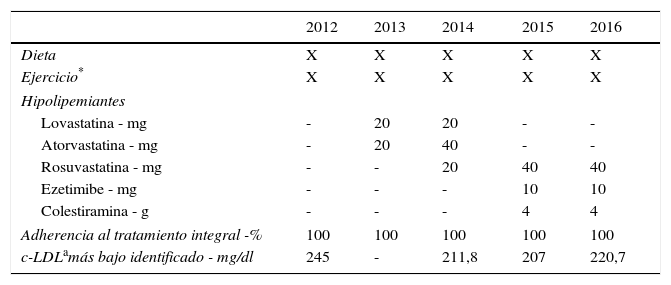
Respecto al tratamiento de su dislipidemia, previo al diagnóstico genético recibió diversos manejos: inicialmente fue tratada con dieta, ejercicio aeróbico (300 minutos por semana de moderada intensidad, según las indicaciones de la Sociedad Americana del Corazón2) y tratamiento farmacológico hipolipemiante intensivo (tabla 2), sin cambios significativos en los niveles de c-LDL (valores de LDL por encima de 200mg/dl). No se reportó intolerancia a las estatinas, pero dada la intensidad del tratamiento se hicieron mediciones de creatinina quinasa (CK), que arrojaron resultados dentro de los límites normales.
Cronología de tratamientos farmacológicos y no farmacológicos recibidos por la paciente
| 2012 | 2013 | 2014 | 2015 | 2016 | |
|---|---|---|---|---|---|
| Dieta | X | X | X | X | X |
| Ejercicio* | X | X | X | X | X |
| Hipolipemiantes | |||||
| Lovastatina - mg | - | 20 | 20 | - | - |
| Atorvastatina - mg | - | 20 | 40 | - | - |
| Rosuvastatina - mg | - | - | 20 | 40 | 40 |
| Ezetimibe - mg | - | - | - | 10 | 10 |
| Colestiramina - g | - | - | - | 4 | 4 |
| Adherencia al tratamiento integral -% | 100 | 100 | 100 | 100 | 100 |
| c-LDLamás bajo identificado - mg/dl | 245 | - | 211,8 | 207 | 220,7 |
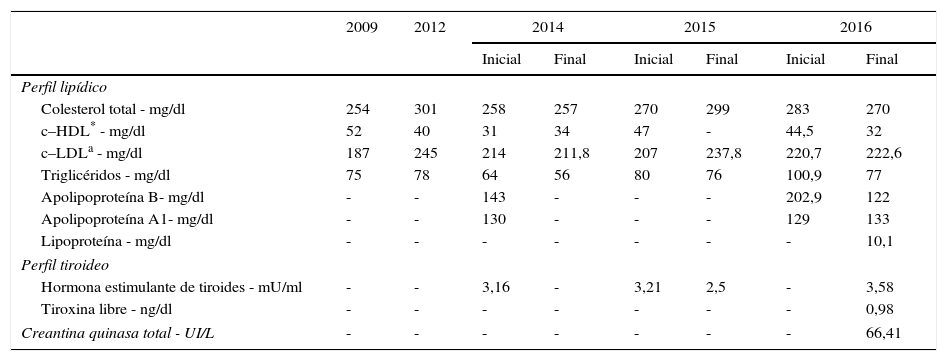
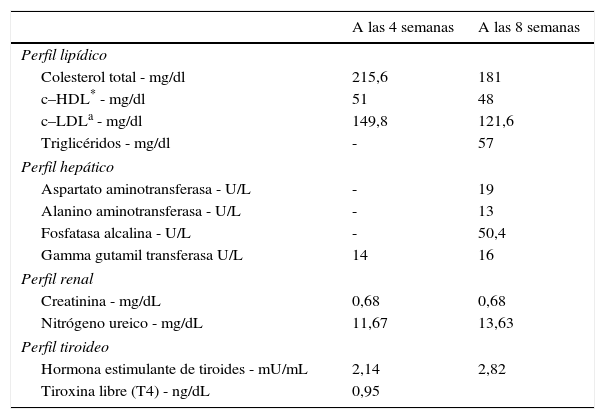
Pese a todas las medidas farmacológicas y no farmacológicas mencionadas, no se logró controlar su perfil de lípidos con la medicación de uso común: estatinas de alta intensidad, tratamiento combinado con ezetimibe y colestiramina (tabla 3). En consecuencia, se decidió iniciar manejo con evolocumab 420mg subcutáneo con frecuencia mensual. Se ordenaron paraclínicos básicos previos al inicio de dicho medicamento para documentar la existencia o no de enfermedades reumatológicas, incluidos anticuerpos nucleares extractables (ENA), anticuerpos antinucleares (ANAS), proteína C reactiva y factor reumatoide, todos estos con resultados negativos. En la tabla 4 se muestra la evolución de los valores de laboratorio encontrados en la paciente a partir del inicio de evolocumab. Cabe anotar que hasta la fecha ha recibido 5 dosis de 210mg quincenales, y no ha presentado eventos adversos; así mismo ha permanecido en estrecho seguimiento por parte del grupo de CES Cardiología.
Exámenes de laboratorio antes del inicio de los inhibidores del PCSK9
| 2009 | 2012 | 2014 | 2015 | 2016 | ||||
|---|---|---|---|---|---|---|---|---|
| Inicial | Final | Inicial | Final | Inicial | Final | |||
| Perfil lipídico | ||||||||
| Colesterol total - mg/dl | 254 | 301 | 258 | 257 | 270 | 299 | 283 | 270 |
| c–HDL* - mg/dl | 52 | 40 | 31 | 34 | 47 | - | 44,5 | 32 |
| c–LDLa - mg/dl | 187 | 245 | 214 | 211,8 | 207 | 237,8 | 220,7 | 222,6 |
| Triglicéridos - mg/dl | 75 | 78 | 64 | 56 | 80 | 76 | 100,9 | 77 |
| Apolipoproteína B- mg/dl | - | - | 143 | - | - | - | 202,9 | 122 |
| Apolipoproteína A1- mg/dl | - | - | 130 | - | - | - | 129 | 133 |
| Lipoproteína - mg/dl | - | - | - | - | - | - | - | 10,1 |
| Perfil tiroideo | ||||||||
| Hormona estimulante de tiroides - mU/ml | - | - | 3,16 | - | 3,21 | 2,5 | - | 3,58 |
| Tiroxina libre - ng/dl | - | - | - | - | - | - | - | 0,98 |
| Creantina quinasa total - UI/L | - | - | - | - | - | - | - | 66,41 |
Exámenes de laboratorio posteriores al inicio de los inhibidores del PCSK9 (evolocumab)
| A las 4 semanas | A las 8 semanas | |
|---|---|---|
| Perfil lipídico | ||
| Colesterol total - mg/dl | 215,6 | 181 |
| c–HDL* - mg/dl | 51 | 48 |
| c–LDLa - mg/dl | 149,8 | 121,6 |
| Triglicéridos - mg/dl | - | 57 |
| Perfil hepático | ||
| Aspartato aminotransferasa - U/L | - | 19 |
| Alanino aminotransferasa - U/L | - | 13 |
| Fosfatasa alcalina - U/L | - | 50,4 |
| Gamma gutamil transferasa U/L | 14 | 16 |
| Perfil renal | ||
| Creatinina - mg/dL | 0,68 | 0,68 |
| Nitrógeno ureico - mg/dL | 11,67 | 13,63 |
| Perfil tiroideo | ||
| Hormona estimulante de tiroides - mU/mL | 2,14 | 2,82 |
| Tiroxina libre (T4) - ng/dL | 0,95 | |
La hipercolesterolemia familiar es un desorden genético autosómico dominante que se caracteriza por niveles de c-LDL elevados en plasma, lo que ocasiona mayor riesgo para padecer enfermedad coronaria aterosclerótica prematura1,3. Este trastorno fue reconocido por primera vez en 1938 por el noruego Carl Muller, quien evidenció que existía una relación entre el nivel de c-LDL, los xantomas tendinosos y las lesiones coronarias4.
Como consecuencia del aumento del c-LDL circulante se generan unos cambios del endotelio que llevan a lesiones ateroscleróticas, enfermedad coronaria temprana, enfermedad arterial periférica y enfermedad valvular (principalmente estenosis aórtica)5,6, además de acumulación de colesterol en la piel, que conduce a la formación de xantomas, particularmente en superficies tendinosas (aquiliana y extensor de los dedos), arco presenil por depósitos en la córnea y xantelasmas por depósitos de colesterol alrededor de los ojos7,8. Los xantomas son patognomónicos de la enfermedad y deben hacer sospechar el diagnóstico a cualquier edad7,9; por su parte, los xantelasmas se asocian más con enfermedad coronaria y mortalidad, de manera independiente del nivel de colesterol plasmático10.
Dentro de la hipercolesterolemia familiar se describen dos presentaciones según su alteración genética y cigocidad: hipercolesterolemia familiar heterocigota y homocigota. En la primera se clasifican niveles de colesterol entre 350-550mg/dl, que se han relacionado con la aparición de enfermedad coronaria en hombres menores de 55 años y mujeres menores de 60 años7,11,12. En la segunda, los niveles de c-LDL alcanzan valores entre 650 y 1.000mg/dl13 y se relacionan con muerte por causas cardiovasculares en menores de 30 años.
Adicionalmente, se ha estimado que estos pacientes pueden padecer su primer evento coronario 20 años antes que la población general (42 años vs. 64 años)14. En un análisis de pacientes con enfermedad heterocigota hecho por el grupo “Simon Broome” en 1980, “era preestatinas”, se documentó un aumento de mortalidad de hasta cien veces, a causa de enfermedad coronaria en jóvenes entre 20-39 años vs. población general15.
En cuanto a su epidemiología, se ha calculado que entre 14 a 34 millones de personas en el mundo sufren hipercolesterolemia familiar, no obstante, a menos del 1% se les realiza un diagnóstico adecuado1. La prevalencia de la forma heterocigota en europeos es de 1 en 500 (0,20%)13, sin embargo, un estudio más reciente de 69.016 individuos con hipercolesterolemia, familiar sugiere una prevalencia mayor: 1 por cada 200 habitantes16. Se han reconocido poblaciones con mayor prevalencia, entre estos, africanos1,7, canadienses, franceses (1 en 270), libaneses (1 en 85) y judíos Ashkenazi (1 en 72)14,17. Por su parte, la forma homocigota de la enfermedad afecta 1 de cada 1′000.000 de personas17.
Según la etiología, se sabe que esta enfermedad envuelve tres mutaciones genéticas diferentes: del R-LDL, de la ApoB y de la PCSK918. En última instancia, todas estas alteraciones genéticas generan disminución de la degradación del c-LDL plasmático mediante diferentes mecanismos. Así, por ejemplo, los R-LDL, que están en su mayoría en la superficie hepática, son responsables de remover el c-LDL circulante. Se han documentado más de 1.288 mutaciones asociadas a este receptor, 79% de las cuales pueden generar hipercolesterolemia familiar, lo que constituye la etiología más común de la enfermedad19.
De otra parte, la ApoB es una proteína que actúa como ligando entre el c-LDL y el R-LDL; si hay mutaciones en la misma se impide la unión y, por tanto, su degradación. Esta mutación es causa del 5% de las hipercolesterolemias familiares.
El PCSK9 es el encargado de la eliminación de los R-LDL por medio de los lisosomas. Las mutaciones que aumentan la acción de esta proteína llevan a niveles disminuidos del R-LDL en los hepatocitos y en consecuencia a hiperlipidemia. Esta mutación contribuye al 1% de las hipercolesterolemias familiares7,20.
En el caso expuesto, las tres principales mutaciones encontradas se relacionan con enfermedades que claramente aumentan el riesgo cardiovascular:
La mutación en el gen ApoA5 (cromosoma 11q23.3) se asocia con hipertrigliceridemia familiar e hiperlipoproteinemia familiar tipo 521. La mutación en el gen ApoE, ubicado en el cromosoma 19q13.32, se relaciona con aterosclerosis e hiperlipoproteinemia familiar tipo 322, mientras que la mutación del SCARB1, cromosoma 12q24.31, se asocia con niveles bajos de proteínas de alta densidad (c-HDL)23.
Hasta la fecha la hipercolesterolemia familiar es subdiagnosticada y, por ende, subtratada. Se estima que solo se reconoce el 20% de los casos13. Un diagnóstico adecuado debe incluir, por tanto, una combinación entre historia familiar, signos clínicos y concentración de c-LDL. Así mismo, es pertinente excluir todas las causas secundarias de hiperlipidemia (diabetes mellitus, hipotiroidismo, enfermedad hepática y renal, medicamentos, sedentarismo, entre otras)4. Para ello existen dos herramientas diagnósticas frecuentemente utilizadas: los criterios de “Dutch Lipid Clinic Netwok” y los de “Simon Broome”, ambos combinan el nivel de LDL en plasma, historia familia y marcadores genéticos24.
Otra forma de diagnóstico es la tamización con un individuo como caso índice, necesaria debido a la alta prevalencia, mortalidad y morbilidad de la enfermedad; es de anotar que es más efectiva cuando se realiza en familiares en primer grado de consanguinidad del caso índice25. Los casos índices deben ser sometidos a estudios genéticos, y la mutación hallada es la que se estudia en los familiares. Este método identifica el 50% de los casos totales de hipercolesterolemia familiar26. Se debe aclarar que hasta un 40% de pacientes diagnosticados con criterios clínicos no presentan una mutación genética identificable; en consecuencia, estos individuos tienden a presentar menores niveles de c-LDL y a tener mejor pronóstico24,27.
El objetivo principal del tratamiento de la hipercolesterolemia familiar es la reducción de los eventos cardiovasculares y la mortalidad. Hasta la fecha, éste se basa en los niveles de c-LDL y no en las mutaciones genéticas específicas3. La Asociación Americana de Lípidos (NLA) y el Instituto Nacional para la Salud y Excelencia Clínica (NICE) del Reino Unido, recomiendan una reducción de la concentración de c-LDL mayor al 50% del nivel previo al tratamiento13,28.
Dentro de las opciones farmacológicas, la primera línea de tratamiento para la hipercolesterolemia familiar heterocigota son las estatinas de alta intensidad (atorvastatina 80mg/día o la rosuvastatina 40mg/día)29, las cuales disminuyen la enfermedad coronaria hasta un 80% si se inician como tratamiento preventivo en la adultez temprana30. La respuesta inicial debe ser monitorizada uno a tres meses después del inicio. Si el objetivo de disminución de más del 50% del valor inicial de c-LDL no se ha logrado después de tres meses, el paso a seguir es la adición de un segundo medicamento, en cuyo caso el más usado es el ezetimibe, que disminuye los niveles de c-LDL un 15-20% adicional y aumenta la cantidad de R-LDL en la superficie hepática3,4,31. Si pese a la doble terapia, tres meses después, el objetivo no se cumple, se agrega un tercer medicamento (PCSK9 o secuestrador de sales biliares)3.
En pacientes con intolerancia a las estatinas, la combinación entre ezetimibe, niacina, secuestradores de ácidos biliares y posiblemente inhibidores PCSK9 representa la única alternativa de tratamiento24. Contrario a lo anterior, un estudio en 1.249 pacientes con hipercolesterolemia familiar heterocigota evidenció que de 96% de los casos tratados con estatinas, solo el 47% lograba la meta (reducción del 50% del c-LDL inicial) y sólo el 21% alacanzaba niveles de c-LDL menores de 100mg/dl. Casi un tercio (27%) de los pacientes que no cumplían las metas ya tenían prescripción de terapia combinada con ezetimibe4,32. Por todo ello, en los últimos 5 años se han intentado crear nuevas tecnologías en busca de medicamentos que controlen de manera más eficiente la hipercolesterolemia familiar y sus complicaciones subsecuentes. En 2012 y 2013, respectivamente, surgieron el lomitapide (inhibidor de la proteína de transferencia de triglicéridos microsomal) y el mipomersen (inhibidor de oligonucleótido antisentido de Apo B100) para hipercolesterolemia familiar homocigota. En 2015 se aprobaron dos inhibidores de PCSK9 (alirocumab y evolocumab) para el tratamiento de la hipercolesterolemia familiar heterocigota33.
Los efectos clínicos del PCSK9 fueron reconocidos inicialmente por Abifadel et al., quienes en 2003, describieron en dos familias francesas mutaciones que potencian la función del gen de PCSK9. Estos pacientes tenían c-LDL elevado en asociación con aumento de enfermedad coronaria34. Posteriormente, estudios en animales identificaron la función del PCSK9, molécula sintetizada en el hígado que en circulación se une al R-LDL hepático y al complejo PCSK9/R-LDL, que es endocitado e internalizado en el lisosoma donde es sometido a degradación. Este proceso reduce la capacidad de remover el c-LDL circulante y se traduce en niveles aumentados del mismo35,36. El “Dallas Heart Study” encontró individuos con mutaciones que disminuían la función del gen PCSK9 llevando a 28% menos c-LDL circulante que la población general37. Estos estudios plantearon la posibilidad de que la inhibición farmacológica del PCSK9 podría disminuir el c-LDL en pacientes con hipercolesterolemia.
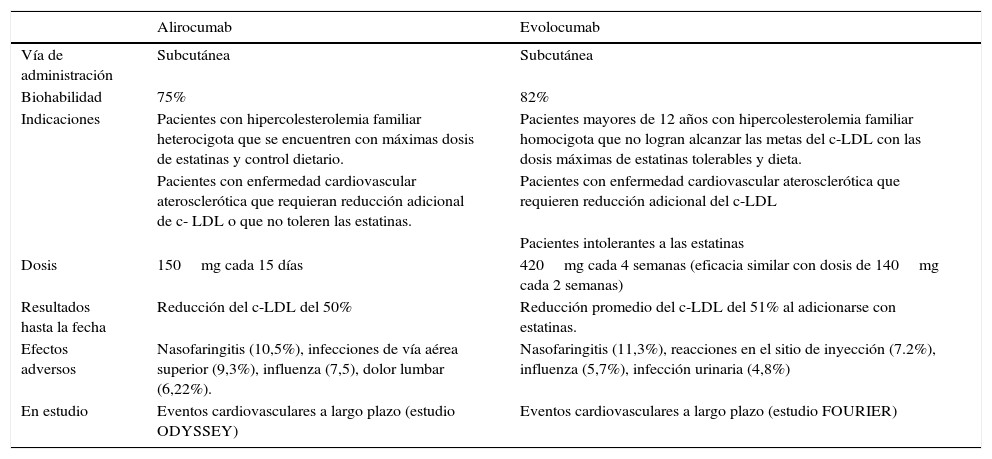
Así pues, se ha logrado la inhibición del PCSK9 mediante la creación de anticuerpos monoclonales humanizados que no atraviesan la barrera hematoencefálica y que son capaces de aumentar la remoción del c-LDL circulante33. Al ser inyectado el anticuerpo anti-PCSK9 se une al PCSK9 circulante agotándolo rápidamente, y en consecuencia se genera menor degradación de R-LDL en compartimentos lisosomales, aumento de R-LDL en la superficie hepática y disminución de los niveles de c-LDL35. Se han creado tres medicamentos de este tipo: alirocumab, evolocumab y bococizumab. Los dos primeros ya están aprobados por la FDA y la EMA para uso en seres humanos. En la tabla 5 se resumen algunas características de estos dos medicamentos38,39.
Características de los inhibidores del PCSK9 aprobados para aplicación en seres humanos
| Alirocumab | Evolocumab | |
|---|---|---|
| Vía de administración | Subcutánea | Subcutánea |
| Biohabilidad | 75% | 82% |
| Indicaciones | Pacientes con hipercolesterolemia familiar heterocigota que se encuentren con máximas dosis de estatinas y control dietario. | Pacientes mayores de 12 años con hipercolesterolemia familiar homocigota que no logran alcanzar las metas del c-LDL con las dosis máximas de estatinas tolerables y dieta. |
| Pacientes con enfermedad cardiovascular aterosclerótica que requieran reducción adicional de c- LDL o que no toleren las estatinas. | Pacientes con enfermedad cardiovascular aterosclerótica que requieren reducción adicional del c-LDL | |
| Pacientes intolerantes a las estatinas | ||
| Dosis | 150mg cada 15 días | 420mg cada 4 semanas (eficacia similar con dosis de 140mg cada 2 semanas) |
| Resultados hasta la fecha | Reducción del c-LDL del 50% | Reducción promedio del c-LDL del 51% al adicionarse con estatinas. |
| Efectos adversos | Nasofaringitis (10,5%), infecciones de vía aérea superior (9,3%), influenza (7,5), dolor lumbar (6,22%). | Nasofaringitis (11,3%), reacciones en el sitio de inyección (7.2%), influenza (5,7%), infección urinaria (4,8%) |
| En estudio | Eventos cardiovasculares a largo plazo (estudio ODYSSEY) | Eventos cardiovasculares a largo plazo (estudio FOURIER) |
El evolocumab es un anticuerpo monoclonal humano completo tipo IgG2. Los estudios hechos hasta el momento estiman que este fármaco logra reducir en un 60% la concentración de c-LDL cuando se administra en las dosis probadas por estudios de fase III40–42, con una disminución de los niveles circulantes de PCSK9 del 85-95% después de una semana de la administración43.
El primer estudio de fase III que evalúo el evolocumab en pacientes con hipercolesterolemia familiar heterocigota fue el RUTHERFORD–2, multicéntrico, doble ciego, aleatorizado, placebo controlado, en el que se incluyeron 331 pacientes: un grupo recibió 210mg de evolocumab cada 2 semanas (420mg mensuales), y el otro placebo. Luego de 12 semanas, el grupo que había recibido dosis quincenales de evolocumab presentaba una disminución del c-LDL entre el 59-60% comparado con el placebo. También se demostró una disminución del 15% en los triglicéridos y un incremento del 7% en el c-HDL. Al parecer, la respuesta al evolocumab es independiente del tipo de mutación que cause la hipercolesterolemia familiar heterocigota42. Para el caso expuesto se calcularon los valores porcentuales en los que aumentaron o disminuyeron los parámetros básicos del perfil lipídico respecto a los valores inmediatamente previos al inicio de la aplicación de evolocumab (tabla 6).
Existen reportes de efectos adversos generados por la administración del evolocumab. En los estudios de fase II y III se informa un abandono al tratamiento entre el 1,9 al 2,3% por eventos adversos44. Los más reportados hasta la fecha son: nasofaringitis (5,9% en el grupo de evolocumab vs. 4,8% en el grupo control), infecciones de vía respiratoria superior (3,2 vs. 2,7%), cefalea (3,0 vs. 3,2%), dolor de espalda (3,0 vs. 2,7%) y mialgias (2,5 vs. 2,6%). Entre los efectos adversos de interés se encuentran reacciones en el sitio de punción (3,3 vs. 3,0%), elevación de creatinina quinasa más de 5 veces el límite superior de normalidad (0,7 vs. 0,7%), elevaciones de ALT y/o AST más de 3 veces al límite superior de normalidad (0,4 vs. 1%) y alteraciones neurocognitivas (amnesia, delirium, desorientación y alteraciones en la memoria) (0,1 vs. 0,3%) las cuales están en estudio en el EBBINGHAUS44,45. Entre 2-8% de los pacientes en los estudios descontinuaron el medicamento, sin claridad sobre la causa de ésta. Así mismo, por la composición del inhibidor de PCSK9 (anticuerpo humanizado) se pueden generar anticuerpos contra el inhibidor de PCSK9, pero hasta ahora no se han demostrado con frecuencia y no han disminuido la eficacia del medicamento33.
ConclusionesPese a que la hipercolesterolemia familiar no se presenta en un gran porcentaje de la población, merece ser sospechada y diagnosticada, a raíz de las consecuencias cardiovasculares significativas que genera. Por tanto, su detección temprana permite impactar en intervenciones oportunas.
En la actualidad, los inhibidores del PCSK9 se han convertido en una alternativa prometedora para aquellos que no responden a las terapias convencionales y cursan con enfermedades tan severas como la hipercolesterolemia familiar. Por ahora, dichos inhibidores, especialmente, el evolocumab en el tratamiento de la hipercolesterolemia familiar heterocigota, parecen ser una terapia prometedora en términos de disminución de valores de colesterol total, c-LDL y triglicéridos y aumento de c-HDL. Faltan estudios y seguimientos muy estrechos y objetivos en el tiempo, que corroboren o contradigan los beneficios y eventos adversos encontrados hasta el momento en los pacientes que se someten a estas nuevas opciones terapéuticas.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónNinguna.
Conflicto de interesesNinguno.