


El síndrome de Cushing es una enfermedad muy rara pero asociada a una morbimortalidad significativa. Se clasifica como dependiente de la hormona adrenocorticotrópica (ACTH: tumores hipofisiarios y ectópicos) o independiente de ACTH (lesiones de origen adrenal). En la mayoría de los casos, las lesiones responsables del síndrome corresponden a tumores hipofisiarios, seguida de lesiones adrenales y por último de tumores ectópicos (5-15% de todos los casos). En este artículo se hará una revisión de los aspectos epidemiológicos, clínicos, diagnósticos y terapéuticos más importantes de los tumores ectópicos causantes del síndrome de Cushing.
Cushing's syndrome is a very rare disease associated with significant morbidity and mortality. It is classified as adrenocorticotropic hormone (ACTH) dependent (Pituitary and ectopic tumors) or ACTH independent (lesions of adrenal origin). In most cases, pituitary tumors are responsible for the Cushing‘s syndrome, followed by adrenal lesions and finally by ectopic tumors (5-15% of all cases). This article describes the most important epidemiological, clinical, diagnostic and therapeutic aspects of ectopic tumors causing Cushing's syndrome.
El síndrome de Cushing (SC) es una entidad muy rara. Previamente se reportó una incidencia de 1 caso por cada 13 millones de habitantes1. Sin embargo, recientemente se ha reportado una incidencia de 2-3 casos por millón de habitantes por año, que probablemente sea mucho mayor, alrededor de 5-25 casos por millón de habitantes por año, ya que hasta el 10% de los incidentalomas adrenales pueden sintetizar cortisol en exceso2. El SC se clasifica como dependiente de la hormona adrenocorticotropa (ACTH), de origen hipofisiario (80-85%) o ectópico (15-20%), e independiente de la ACTH, de origen adrenal. En el 5 al 15% de todos los casos el SC se debe a un tumor ectópico (tumores neuroendocrinos (TNE) y algunos tumores de órganos sólidos)3,4. En esta revisión se tratarán los aspectos más relevantes del síndrome de Cushing ectópico (SCE).
MétodosPara esta revisión se realizó una búsqueda con los términos MESH “ACTH syndrome, ectopic”, “paraneoplastic endocrine syndrome” y “Cushing syndrome” en inglés en las bases de datos Medline, Scielo, Lilacs y Bireme. En la búsqueda se incluyeron estudios originados en Colombia: un trabajo original5, dos revisiones de tema6,7 y dos reportes de caso8,9. Se excluyó además un reporte de caso por ser de carcinoma adrenal y una revisión de tema no relacionado con el objetivo de este trabajo.
EpidemiologíaLa edad de presentación es más tardía en comparación con otros casos de SC (hasta 10 años), y en general no hay un predominio tan marcado en las mujeres como sí ocurre en el SC de otras etiologías. Se han descrito relaciones mujer: hombre de 1:1 o 1:2 en las diferentes publicaciones3,10–12
El 40 al 60% de los tumores responsables del SCE se encuentran en el tórax4,11,12. Las neoplasias más relacionadas son los carcinoides bronquiales (25-32%), carcinomas de células pequeñas del pulmón (20%), TNE del timo (7-11%) y de las células de los islotes pancreáticos (8%), feocromocitomas (3,4-5%) y carcinoma medular de tiroides (0,6-7,5%). También se han descrito casos de SCE en tumores sin diferenciación neuroendocrina (6-8% de los casos): adenocarcinoma de ovario, cérvix, anorectal, pulmón, próstata y neuroblastoma4,8,11–18. Los TNE se caracterizan por la producción de una o varias hormonas (gastrina, catecolaminas, serotonina, calcitonina, péptido intestinal vasoactivo, bombesina, entre otras) que causan diferentes síndromes clínicos. Además pueden producir varios marcadores neuroendocrinos (cromogranina A, sinaptofisina, ácido 5 hidroxi indol acético) que pueden orientar al diagnóstico de TNE como ocurre por ejemplo con la cromogranina A10,13,18. La hormona que se produce con más frecuencia en el SCE es la ACTH, y en una proporción mucho menor la hormona estimulante de corticotrópos (CRH). Se ha descrito la secreción concomitante de ambas hormonas19, y en un caso se encontró un TNE del timo con expresión de ACTH al que se le hizo manejo quirúrgico del tumor primario con resolución del hipercortisolismo, pero en una recaída posterior del SCE a los 6 años la hormona que se identificó fue la CRH20.
En un estudio retrospectivo realizado entre 1986 y 2010 en el Hospital San Vicente Fundación (HSVF) de Medellín (Colombia) 5 se encontraron 30 casos de SC, de los cuales 4 eran de origen ectópico (dos tumores neuroendocrinos: bronquial y pancreático, un carcinoma medular de tiroides con expresión de ACTH y un tumor no identificado - oculto), correspondientes al 13% del total de la muestra. Los pacientes tenían una edad promedio de 33,5 años (25-46 años); tres fueron mujeres y uno hombre. Como en esta serie, en algunos casos no se logra identificar la neoplasia responsable del hipercortisolismo, con frecuencias variables en las diferentes publicaciones que oscilan entre el 12,5% al 38%3,10–12,21. Sin embargo, es muy probable que en la actualidad esta cifra sea mucho menor debido a la mejoría significativa en los estudios de localización18.
ClínicaLa liberación de estas hormonas puede causar un SC con los hallazgos clínicos característicos de esta entidad, resaltando la hipertensión y alteración en el metabolismo de la glucosa, la debilidad y atrofia muscular proximal, la hipocalemia (en más del 70% de los casos), la fragilidad capilar e hiperpigmentación cutánea3,4,7,11–13,18,22,23. En muchas circunstancias los síntomas son muy graves y de rápida instauración como ocurre en los tumores de células pequeñas del pulmón y otros TNE bronquiales, mientras que en otros casos el comportamiento es insidioso como ocurre en los TNE de otros órganos3,13,18,22,23. En el SCE se han descrito alteraciones psiquiátricas, osteoporosis, infecciones y fracturas hasta en el 50% de los afectados. También se ha reportado un aumento en la incidencia de enfermedad tromboembólica10,12,18,24. En los casos de rápido inicio de la enfermedad los pacientes pueden tener ausencia de signos clásicos del SC como las facies de luna llena y la obesidad centrípeta y pueden tener hiperpigmentación marcada18, y muchos pueden perder peso en vez de ganarlo10,12. Los síntomas de hipercortisolismo pueden ser la manifestación inicial o pueden aparecer varios meses e incluso años después del descubrimiento del tumor primario3,18. En la serie del HSVF 5 se encontró en todos los pacientes diabetes, HTA, dislipidemia, obesidad centrípeta y estrías gruesas y pigmentadas en flancos; dos de las pacientes tenían además osteoporosis, y una de ellas tuvo múltiples fracturas vertebrales. Tres de los cuatro pacientes tuvieron hipocalemia, la cual fue de difícil corrección. Ninguno tuvo hiperpigmentación. Una paciente con carcinoma medular tenía metástasis hepáticas y presentó un cuadro de diarrea crónica y flushing5,8.
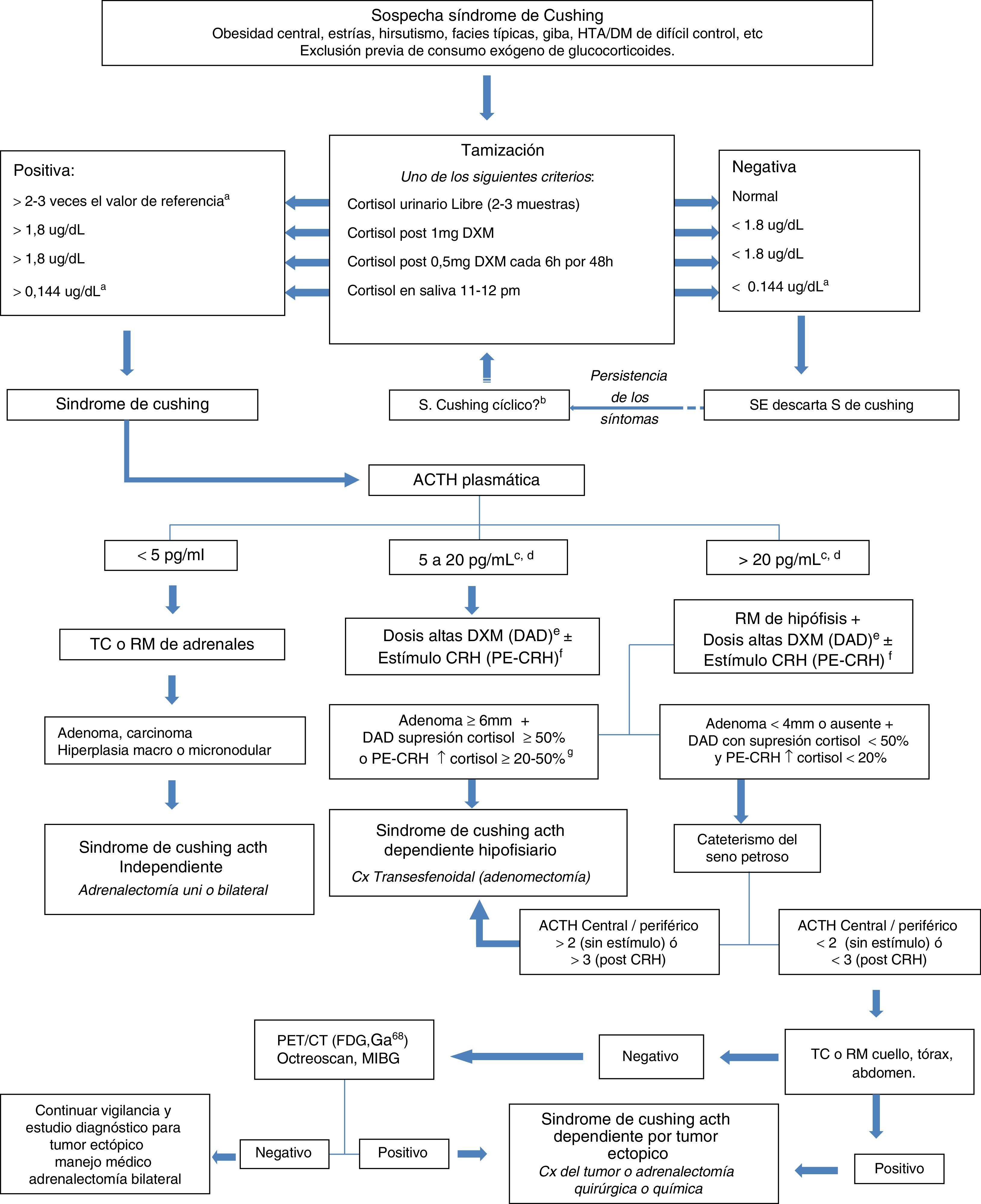
DiagnósticoEn la figura 1 se anexa el algoritmo diagnóstico utilizado para el diagnóstico del SC y sus variantes7. En el SCE, el valor del cortisol urinario libre de los metabolitos del cortisol y las concentraciones de ACTH son mucho mayores con respecto al SC causado por adenomas hipofisiarios7,11,13,14,18,23, aunque también se ha descrito que los valores de ACTH se pueden sobreponer con los de pacientes con enfermedad de Cushing3. En el SC de origen ectópico, se debe tener en cuenta que hasta el 10% de la población normal puede tener incidentalomas hipofisiarios, por lo que en muchas ocasiones se deben hacer pruebas dinámicas complementarias para poder hacer un diagnóstico diferencial apropiado y evitar cirugías transesfenoidales innecesarias3,4,25.
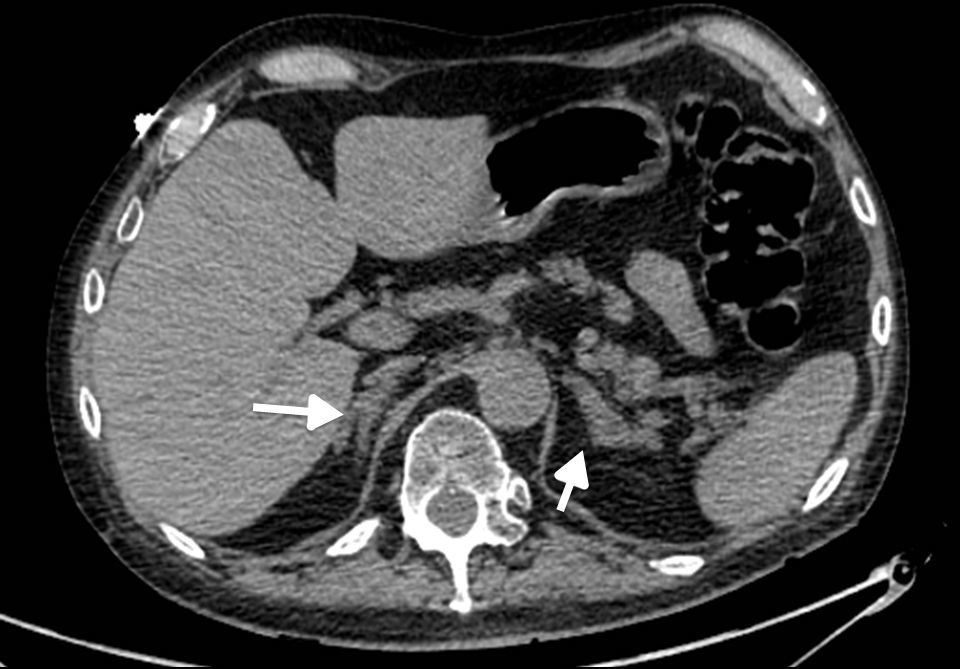
 con metástasis hepáticas y síndrome de Cushing. Se aprecia hiperplasia suprarrenal bilateral (flechas).")
El principal diagnóstico diferencial del SCE es la enfermedad de Cushing7,18. Se ha demostrado que los tumores hipofisarios son los menos autónomos de los causantes del SC, y que las dosis altas de esteroides (p.e dexametasona) suprimen la secreción de cortisol en adenomas hipofisarios (en más del 50% tras una dosis de 8mg), mientras que en la mayoría de los tumores ectópicos (90%) no suprime7,26,27. En la serie del HSVF la ACTH promedio fue de 130,5pg/ml y los valores de cortisol en orina de 24 horas fueron los más altos de toda la cohorte: 2.975 ug, (VR 20-148). En dos de tres pacientes el cortisol post-dosis altas de dexametasona (8mg) suprimió en más del 50% respecto al basal5.
La mayoría de las lesiones que causan un SCE por producción de ACTH no expresan receptores para CRH, por lo cual el uso de una prueba de estímulo con esta hormona puede ayudar a hacer el diagnóstico diferencial entre estas dos entidades. Para esta prueba se usa CRH ovina (1μg/kg) o humana (100μg), aplicada por vía intravenosa a las 9 AM, seguida de la medición de ACTH y cortisol desde los -15 minutos hasta los 60 a 120 minutos post-aplicación. Se espera un aumento de la ACTH mayor al 35% a los 15 a 30 minutos en los casos de enfermedad de Cushing tras la aplicación de la CRH, con una sensibilidad reportada del 93% y una especificidad del 100%7,26,27. En los casos de SCE no habrá respuesta o el aumento será menor de 1,5 veces con respecto al valor inicial28.
En la enfermedad de Cushing también se puede ver un aumento del cortisol del 14% al 20%, dependiendo de la presentación de CRH usada, con una sensibilidad del 91% y una especificidad del 88%7,26,27. A pesar de lo anterior, se debe tener en cuenta que hasta un 10-15% de los tumores ectópicos pueden responder al estímulo con CRH o suprimir el cortisol con dosis altas de dexametasona, por lo que en casos dudosos se deben combinar las dos pruebas para lograr tener una mejor certeza diagnóstica7,26,27. Si esto no es posible o los resultados son contradictorios, se debe hacer un cateterismo del seno petroso, el cual es el estándar de referencia para hacer el diagnóstico diferencial entre una enfermedad de Cushing y un tumor ectópico, y consiste en la medición de la ACTH que drena directamente de la hipófisis y con base en ello se establece un gradiente de concentración con la ACTH de una vena periférica. El examen se puede hacer también tras la aplicación de CRH, que estimulará a las células del adenoma para producir ACTH. Si se obtiene un gradiente de ACTH seno petroso/vena periférica mayor de 2 sin estímulo, o mayor de 3 con estímulo con CRH es indicativo de una enfermedad de Cushing con una sensibilidad y especificidad cercanas al 100%, y una relación mayor de 1,4 entre un lado de la hipófisis respecto al otro indica el sitio donde se encuentra el adenoma (aunque la sensibilidad para la localización del adenoma no es tan buena y está alrededor del 78%)6,21,29–34. Como alternativa a la CRH (difícil de conseguir en nuestro medio), se puede usar la desmopresina (sensibilidad y especificidad de 92,1% y 100% respectivamente), y la interpretación es igual a como se mencionó con la CRH6. Para confirmar que el cateterismo fue exitoso, se puede hacer una medición de prolactina y calcular el gradiente prolactina seno petroso/vena periférica, donde un valor mayor de 1,8 indica que el catéter se posicionó adecuadamente en el seno petroso6,35. En los estudios realizados a la fecha el cateterismo ha demostrado ser útil para hacer una diferenciación entre enfermedad de Cushing y tumores ectópicos25. Si la relación de ACTH seno petroso/vena periférica es menor de 2 sin estímulo o menor de 3 con estímulo, se debe proceder a la realización de imágenes en tórax, abdomen y cuello para tratar de identificar un tumor ectópico que explique el cuadro clínico6,7,18,34.
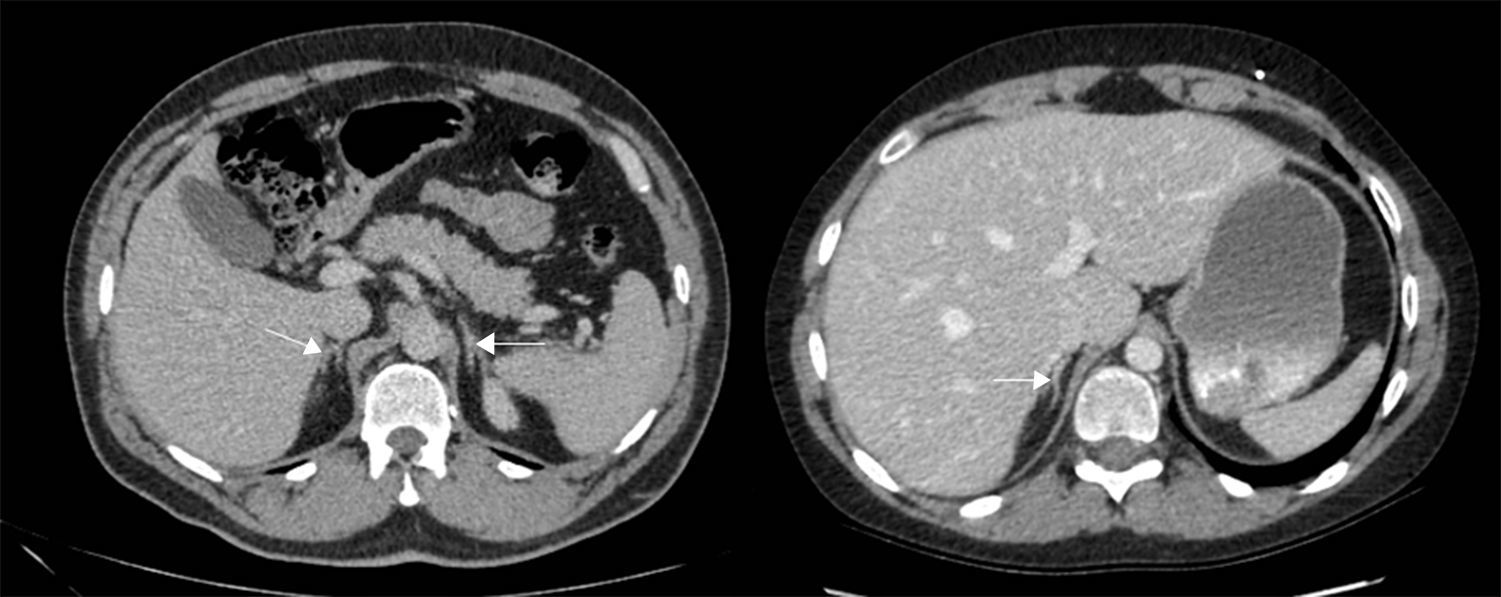
Los estudios de imágenes convencionales (fig. 2) permiten la localización de estos tumores, pero cuando son negativos (lo que puede ocurrir en el 20% de los casos) se pueden usar imágenes de medicina nuclear con Indio111-octreotido, Yodo123-metabencilguanidina (MIBG), entre otros, que permiten el diagnóstico en algunos de los casos no identificados por otros medios de imágenes23,36–39. Sin embargo, al realizar estudios con análogos de somatostatina radiomarcados se debe tener en cuenta que no todos los tumores poseen receptores o los subtipos para los cuales los análogos de somatostatina son más afines (receptores 2 y 5), y pueden existir falsos positivos en patologías no neoplásicas como neumonía, enfermedades granulomatosas o fibrosis postradioterapia 23,36,37 y en varias series de casos de SCE el rendimiento diagnóstico de estas imágenes no ha sido el mejor, con una sensibilidad de tan solo el 50-60%23,36,37. Además en la mayoría de casos en los que la tomografía o la resonancia fueron negativos, el estudio de medicina nuclear también fue negativo10–12. Recientemente se ha venido utilizando la tomografía con emisión de positrones (PET/CT), donde el marcador galio (68Ga-DOTATATE) ha demostrado que puede ser útil en la localización de tumores no detectados en otras imágenes, con una mayor sensibilidad en comparación al marcador tradicional 18 fluorodeoxiglucosa (FDG-PET, 82% vs. 66%) en tumores bien diferenciados4,25,38–40. En tumores de alto grado o indeterminados la combinación de 68Ga-DOTATATE con PET/CT con 18 fluorodeoxiglucosa (FDG) podría mejorar la sensibilidad diagnóstica a un 92%39. También se ha usado el marcador 18F-DOPA con buenos resultados20,41. En estos estudios también se pueden presentar falsos positivos, como en el caso de procesos inflamatorios (colecistitis, enfermedades granulomatosas, cicatrices previas) y falsos negativos (lesiones menores de 1cm, insulinomas benignos, TNE malignos)40. En una revisión sistemática de la literatura42 se comparó la eficacia de diferentes estudios de imágenes en la localización de TNE causantes del SCE: tomografía 66,2%; RM 51,5%; Indio111-octreotido 48,9%; FDG-PET 51,7%; F-DOPA-PET 57,1%; MIBG 30,8%, y Ga68 con análogo de somatostatina-PET 81,8%. Los estudios con galio tuvieron una sensibilidad del 100% para la localización del TNE en SCE con tumores ocultos (figs. 3 y 4).
 de origen pancreático con metástasis hepáticas (flecha gruesa) y síndrome de Cushing con hiperplasia suprarrenal bilateral (flecha delgada).")
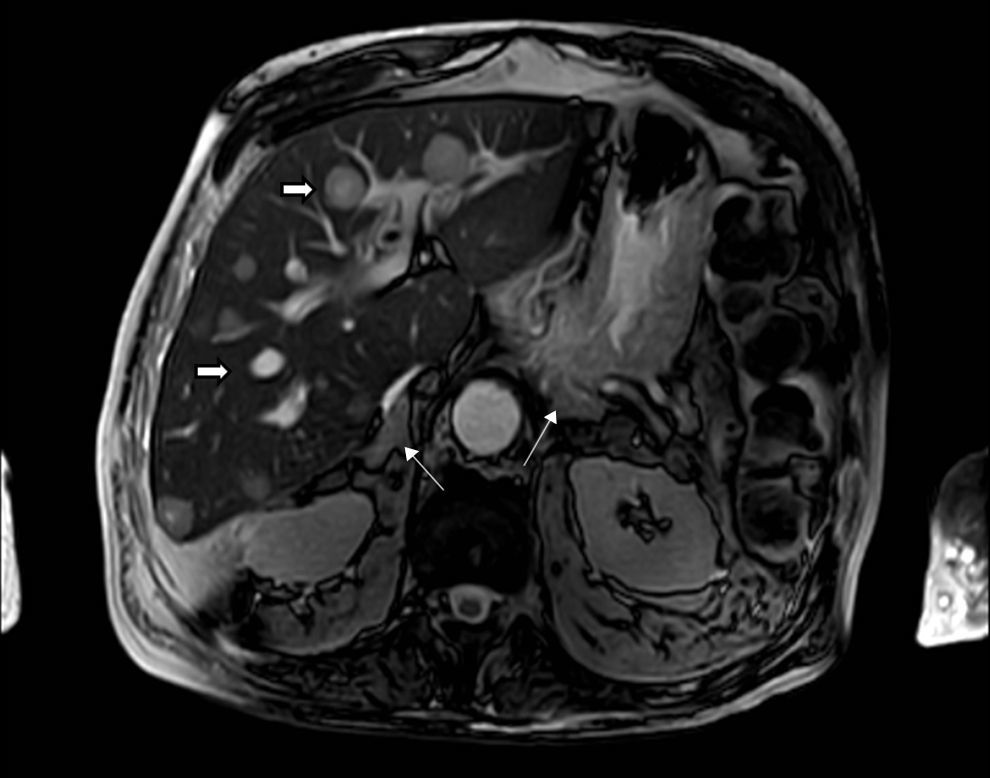
Síndrome de Cushing ectópico por carcinoma neuroendocrino pancreático. Resonancia abdominal de un paciente con carcinoma neuroendocrino (Ki67 40%) de origen pancreático con metástasis hepáticas (flecha gruesa) y síndrome de Cushing con hiperplasia suprarrenal bilateral (flecha delgada).


Algoritmo diagnóstico en el síndrome de Cushing. a. Los valores varían según rangos de referencia del laboratorio. b. Si persiste la sospecha repetir los estudios periódicamente. c. Considerar síndrome de Cushing ACTH dependiente que ha conducido a una hiperplasia adrenal que se ha automatizado. d. se interpreta igual que en los casos con ACTH mayor de 20 pg/mL. e. Si con la prueba con dosis bajas de DXM suprimió más del 50% de la concentración basal de cortisol, se puede omitir. f. Si en la prueba con dosis altas de dexametasona no hay supresión del cortisol mayor del 50% se debe hacer el estímulo con CRH. g. También se puede considerar el aumento de la ACTH del 35-50% tras CRH como prueba positiva.
CX: cirugía; DM: diabetes Mellitus; DXM: dexametasona; HTA: hipertensión arterial; TC: tomografía; RM: resonancia magnética; PET: tomografía por emisión de positrones.
En cuanto al tratamiento, en el SCE se debe hacer manejo quirúrgico de la lesión primaria, y en las distintas series, este procedimiento se asocia a tasas de curación del hipercortisolismo muy variables (12% a 71%) ante los múltiples tipos de tumores que lo pueden causar y que tienen comportamientos biológicos muy diferentes que en muchas ocasiones impiden su resección quirúrgica10–12,15. Las cifras de curación tumoral oscilan entre el 29 al 35%11,25, aunque algunas neoplasias como los tumores neuroendocrinos bronquiales pueden tener mejores resultados (curación en el 72-83% de los casos)5,10,25. La supervivencia reportada es del 32,2% a 35%10–12. La mortalidad también es muy variable en las distintas series debido a los diferentes tipos de tumores identificados en las mismas: 21% a 63%11,12,15,16. En estos pacientes el pronóstico depende del tipo de tumor (por ejemplo, en el carcinoma de células pequeñas de pulmón la sobrevida promedio es de 6-8 meses mientras que en los carcinoides y tumores ocultos es mucho mayor) y de la presencia de metástasis al momento del diagnóstico del SC (como ocurre en el carcinoma medular de tiroides y los TNE de las células de los islotes pancreáticos)8,13,18,23,43,44.
En muchos casos la enfermedad tumoral está muy avanzada y no se puede hacer resección del tumor o el hipercortisolismo es tan grave que se requiere un control rápido del mismo. Para estos pacientes existen otras alternativas como la adrenalectomía bilateral, la cual se asocia a una rápida resolución del hipercortisolismo. Se ha reportado una mejoría significativa tras la cirugía en muchas de las manifestaciones del Cushing como hipertensión, diabetes, debilidad muscular, alteraciones menstruales, obesidad, cambios fenotípicos y calidad de vida45–48, aunque la fatiga y los trastornos psiquiátricos pueden persistir en el tiempo48,49. En general, la adrenalectomía es segura y es una excelente alternativa para pacientes sin adecuada respuesta al tratamiento médico, con cortos tiempos de estancia hospitalaria (3 días) y baja tasa de complicaciones45. La adrenalectomía laparoscópica se asocia a menor incidencia de sangrado, menor estancia hospitalaria y morbilidad respecto a la adrenalectomía abierta, pero no ha demostrado unas menores tasas de mortalidad45–48,50.
La supervivencia de los pacientes operados es mejor al compararlos con pacientes no intervenidos (sobrevida a un año de 67% vs. 0%), en especial con tumores no identificados en el estudio diagnóstico47, y puede llegar a ser hasta 2,5 años mayor en pacientes con TNE del páncreas50. Las tasas de mortalidad tras la adrenalectomía bilateral a 30 días son mayores en los casos de SCE en comparación con la enfermedad de Cushing (4% vs. 1%), siendo las infecciones y sepsis las causas principales. Es probable que esto se deba a una mayor gravedad del hipercortisolismo y de la inmunosupresión y una enfermedad tumoral más avanzada en los primeros. A largo plazo (35 meses), la media de la mortalidad fue del 39%48 y del 44% a 10 años49, principalmente por progresión del SC48.
Otra alternativa es el uso de medicamentos que inhiban la síntesis de glucocorticoides (ketoconazol, metirapona) o que sean adrenolíticos (mitotane), que permiten el control total o parcial del hipercortisolismo (cortisol urinario libre dentro del valor normal de referencia del laboratorio), y se pueden usar de forma conjunta con la quimioterapia u otras modalidades terapéuticas para el manejo de la neoplasia de base, como terapia puente antes de la cirugía u otras intervenciones y como terapia inicial en SCE secundario a tumores ocultos. Estas terapias se asocian a tasas variables de control del hipercortisolismo y pueden causar toxicidad significativa (vómito, diarrea, dolor abdominal, hepatitis aguda, hipogonadismo masculino, alteraciones neurológicas, alargamiento del QT, inducción de insuficiencia adrenal, entre otras) e interacciones farmacológicas (actúan sobre la enzima CYP3A4) que pueden dificultar la tolerancia y el uso a largo plazo11,51–54. Sin embargo, se han descrito algunos casos de pacientes que recibieron tratamiento combinado con estas medicaciones (metirapona y ketoconazol; metirapona, ketonazol y mitotane) con control del hipercortisolismo por varios años con buena tolerancia55–57.
Los tumores productores de ACTH pueden expresar receptores de somatostatina y de dopamina, y al actuar sobre ellos se puede regular la producción de cortisol. Se han reportado casos manejados con análogos de somatostatina en monoterapia (octreotide) 58,59 o en combinación con agonistas del receptor D2 de dopamina (cabergolina)11,60–62 con reportes de control variable del hipercortisolismo pero sin impacto sobre el crecimiento tumoral. A la fecha no hay estudios que evalúen la terapia con pasireotide en SCE. No obstante, existen varios protocolos que evaluarán la utilidad en pacientes con tumores neuroendocrinos con manifestaciones clínicas (clinicaltrials.gov)
En casos de TNE agresivos (carcinomas neuroendocrinos) o con metástasis asociadas se ha utilizado la quimioterapia63. También se ha descrito la terapia con radioisótopos en casos de enfermedad metastásica o no susceptible de resección (principalmente en TNE gastroenteropancreáticos y de pulmón) e inclusive como adyuvancia prequirúrgica con sustancias como itrio (Y90-DOTATOC) y lutecio (Lu177-OCTREOTATE), los cuales se asocian a tasas de control variables (4 a 33%) y medias de sobrevida de 22 a 46 meses37. También se ha encontrado mejoría en la calidad de vida, control sintomático en el 40 a 70% de los casos, y una sobrevida libre de progresión de 17 a 29 meses40,64. Se ha utilizado también la radiofrecuencia o la quimioembolización para el manejo de metástasis hepáticas y de radioterapia en lechos tumorales o en sitios de metástasis11,40. En pacientes con SCE secundario a carcinoma medular de tiroides65 se ha logrado el control del hipercortisolismo con el uso de los inhibidores de tirosina quinasa66 vandetanib y sorafenib67–69.
En la serie del HSVF hubo control completo del hipercortisolismo en los dos pacientes con TNE, tras la resección exitosa del tumor y la adrenalectomía bilateral abierta en la paciente con tumor neuroendocrino pancreático. Sin embargo, la paciente con TNE bronquial presentó una recaída del SC siete años después al presentar recidiva tumoral en el mismo sitio; tras la cirugía volvió a entrar en remisión sin recaídas hasta el último seguimiento disponible en 2013. La paciente con carcinoma medular de tiroides tenía enfermedad metastásica avanzada que impidió la resección tumoral, pero tuvo mejoría de las manifestaciones del SC tras la realización de adrenalectomía bilateral y control parcial de la diarrea crónica y del flushing con un análogo de somatostatina5,8. Posteriormente falleció ante el extenso compromiso tumoral que presentaba8. El paciente con TNE no identificado se manejó con ketoconazol, con el que se logró controlar el hipercortisolismo y mejoró notoriamente el control glucémico y de su hipertensión. Sin embargo, se desconoce su evolución posterior porque se perdió del seguimiento. La hipocalemia se resolvió por completo en todos los pacientes5.
En conclusión, el SCE es una entidad poco frecuente que puede tener manifestaciones clínicas, tasas de supervivencia y pronóstico muy variables dependiendo del tipo de tumor que la cause. Esta entidad representa un gran reto diagnóstico para el clínico y su tratamiento está encaminado a la resección del tumor primario o a su control con terapia sistémica, y en muchas ocasiones se requiere de forma concomitante el manejo del hipercortisolismo con medicamentos que inhiben la síntesis adrenal de glucocorticoides o mediante la realización de adrenalectomía bilateral.
Fuente de financiaciónNinguna.
Ninguna.
