


Los plasmocitomas intracraneales solitarios son más frecuentes en adultos y predominan ligeramente en las mujeres. Esta localización es extremadamente rara y representa menos del 1% de los tumores de cabeza y cuello; son benignos a menos que se asocie con mieloma múltiple. Presentamos el caso de un paciente masculino de 52 años, sin antecedentes patológicos, con síndrome confusional e hipertensión endocraneana, a quien se le diagnosticó meningioma por tomografía, y se realizó exceresis tumoral Simpson I intradural, con duramadre engrosada. El estudio patológico reportó discrasia de células plasmáticas y positivo para CD138 y CD79a y lambda diagnosticándose plasmocitoma, posteriormente se realizaron exámenes para descartar mieloma múltiple. Debido a las similitudes tanto en la clínica como en la neuroimagen que pueden compartir los meningiomas y plasmocitomas, es necesaria una adecuada correlación con el diagnóstico histopatológico e inmunohistoquímico para la toma decisiones puesto que tanto el pronóstico como el tratamiento dependerán del diagnóstico diferencial.
Intracranial solitary plasmacytomas are more common in adults, with a slight predominance in women. This location is extremely rare, and represents less than 1% of head and neck tumours. They are benign unless associated with multiple myeloma. The case is presented on a 52 year-old male patient, with no medical history, with confusional syndrome and intracranial hypertension, who was diagnosed meningioma using computed tomography. The tumour, with thickened dura, was removed using Simpson I procedure. The histopathology study showed dyscrasia, positive plasma cells, and positive for CD138, CD79a, and lambda. A plasmacytoma was diagnosed after tests were performed to rule out multiple myeloma. Due to the similarities, both in clinical and neuroimaging, that can be shared by meningiomas and plasmacytomas, adequate correlation with the histopathology and immunohistochemistry diagnosis is necessary for decision making, since the prognosis and treatment depend on the differential diagnosis.
Las neoplasias de células plasmáticas, según la clasificación más reciente de la organización mundial de la salud (OMS), se pueden dividir en: plasmocitoma extramedular, que incluye tumor maligno de células plasmáticas, plasmocitoma, y mieloma múltiple, también conocido como mieloma de células plasmáticas1.
Los plasmocitomas son neoplasias que se caracterizan por la proliferación monoclonal de células plasmáticas y la producción de inmunoglobulinas. Se pueden presentar como lesiones solitarias o difusas, se consideran lesiones solitarias al plasmocitoma solitario extramedular y al plasmocitoma solitario de hueso, juntos corresponden a menos del 10% de los tumores de células plasmáticas. El mieloma múltiple es la proliferación difusa y maligna de células plasmáticas2.
Los plasmocitomas intracraneales solitarios son más frecuentes en adultos y ligeramente predominantes en las mujeres. La edad media de presentación es a los 55 años, y pueden tener tres localizaciones: los huesos craneales, la duramadre y el parénquima cerebral. La localización intracraneal es extremadamente rara representando menos del 1% de los tumores de cabeza y cuello, muestran un curso benigno a menos que se asocien con mieloma múltiple1,3–6. La incidencia del plasmocitoma solitario intracraneal es de aproximadamente 3/1.000.000 anual, habiéndose descrito solo 37 casos en la literatura en inglés (hasta el 2014)4. La afección meníngea del mieloma múltiple es muy rara y más extraño es la presencia del plasmocitoma extramedular en esta región7,8.
El principal diagnóstico diferencial consiste en separar un plasmocitoma solitario de una afección dural por mieloma múltiple, ya que el pronóstico y tratamiento en ambos casos es completamente diferente. Es más probable que evolucionen a mieloma los plasmocitomas óseos que los meníngeos y los primeros pueden progresar a mieloma múltiple en un período que varía de 3 a 23 años5. El diagnóstico final se hace correlacionando los hallazgos patológicos con la evaluación sistémica, para descartar mieloma múltiple3. Debido a que se trata de una presentación poco común de tumor cerebral y la importancia que tiene el diferenciar entre meningioma y plasmocitoma para el tratamiento y pronóstico del paciente, se reporta este caso clínico.
Presentación de caso clínicoPaciente de género masculino de 52 años de edad, de raza mestiza y procedente de Talanga, Francisco Morazán, zona rural de Honduras. Con cuadro clínico de: cefalea de leve a moderada intensidad, sin predominio de horario; síndrome confusional; mareos; disminución de la agudeza visual de dos meses de evolución; diplopía y prosopagnosia de inicio súbito de dos semanas de evolución, progresiva que impide actividades de rutina, acompañado de un episodio de vómito. También manifestó disminución de la fuerza en ambos miembros inferiores y miembro superior izquierdo de igual tiempo de evolución, sin antecedentes patológicos de importancia.
En el examen físico tuvo presión arterial 150/100mmHg, y el resto de los signos vitales estaban dentro de parámetros normales. En el examen neurológico presentó: Glasgow de 15, funciones corticales normales, al evaluar pares craneales; agudeza visual 20/40 bilateral, al fondo de ojo presentó papiledema bilateral grado II y diplopía, el resto del examen neurológico dentro de los parámetros normales. Se realizó hemograma completo (tiempos de coagulación y química sanguínea; glucosa; pruebas de función renal; transaminasas hepáticas; lactato deshidrogenasa y electrolitos) todos sin alteraciones.
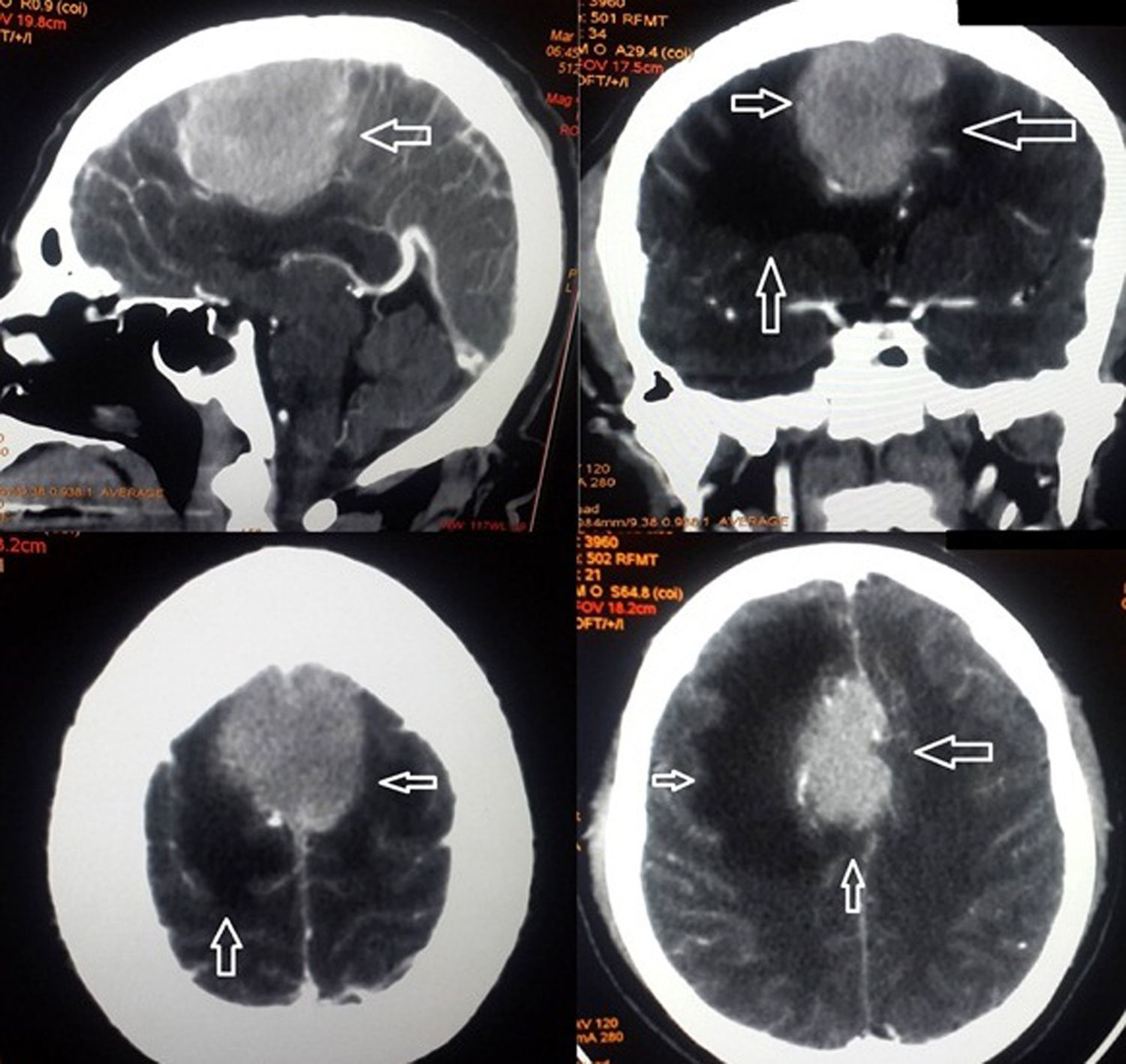
La tomografía computarizada indicó en cortes axiales imagen hiperdensa en la cisura medial, con características de meningioma dependiente de la hoz del cerebro con importante edema pericisural y borramiento del espacio subaracnoideo (fig. 1). Se ingresó con diagnósticos de: síndrome de hipertensión endocraneana, síndrome confusional agudo, edema frontal secundario y probable meningiona; iniciándose manejo con manitol, dexametasona, diclofenaco y fenitoina. El paciente presentó resolución de síndrome confusional y normalización de presión arterial. Quince días posteriores al ingreso se realizó cirugía que consistió en exceresis tumoral Simpson I intradural, con dura madre engrosada. El paciente evolucionó satisfactoriamente, con resolución de síntomas, fue dado de alta al cuarto día postoperatorio.

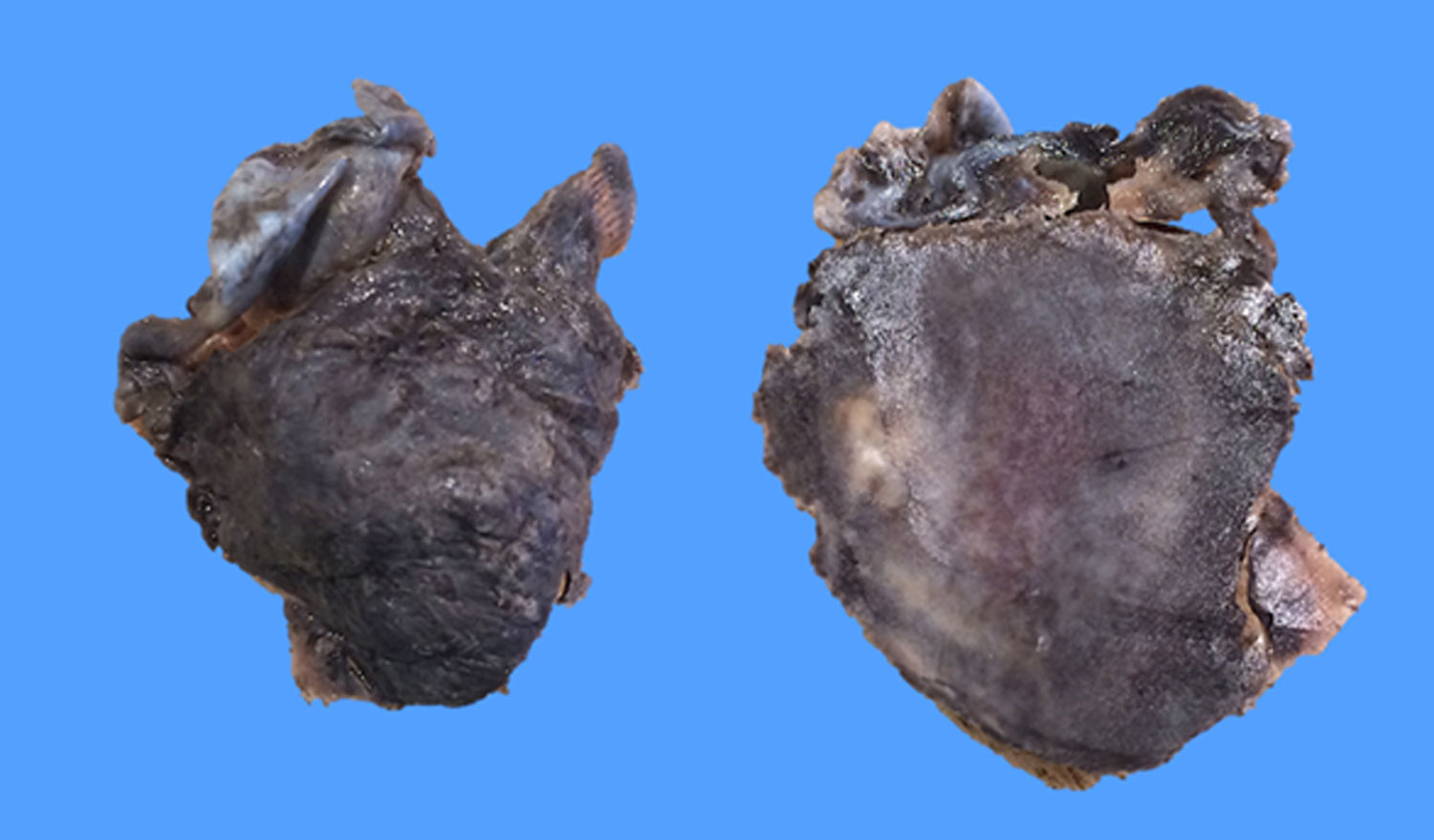
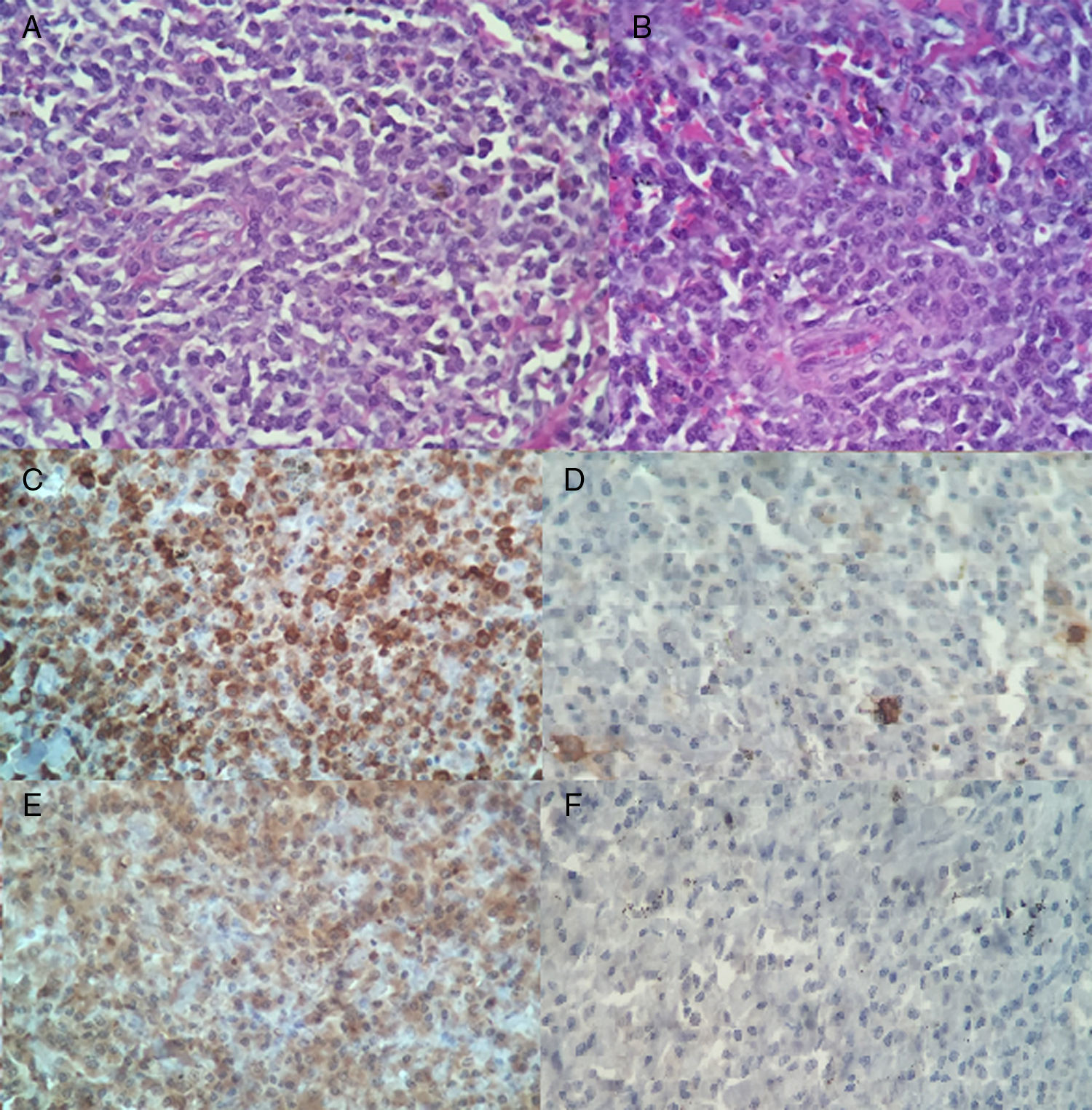
Posteriormente, el estudio histopatológico reportó un fragmento irregular de tejido que medía 3,5×3x2cm, con superficie externa violácea irregular, opaca, en uno de sus extremos adherida estructura de aspecto membranoso de 3×3x0,1cm, grisáceo, opaca, al corte superficie grisácea alternando con áreas violáceas, lisa, opaca (fig. 2). La tinción de hematoxilina eosina mostró discrasia de células plasmáticas (fig. 3 A-B). Inmunohistoquímica positiva para: CD79 (fig. 3C), lambda (fig. 3E), CD138 positivo moderado multifocal, Ki-67 mostró índice de proliferación bajo de 10 a 15%. Negativa para: CD20 (fig. 3D), kappa (fig. 3F), CD34 y EMA (antígeno epitelial de membrana), CD45 resultó negativo para las células tumorales plasmáticas y positivo para linfocitos presentes en la lesión, no se realizó CD56 por no estar disponible en nuestro medio.


A y B discrasia de células plasmáticas en tinción de hematoxilina eosina. C. Marcador CD79a positivo citoplasmático, difuso, intenso. D. CD20 negativo para células plasmáticas y positivas para linfocito B presente. E. Cadena ligera lambda positivo para membrana y citoplasma. F. Cadena ligera kappa negativo.
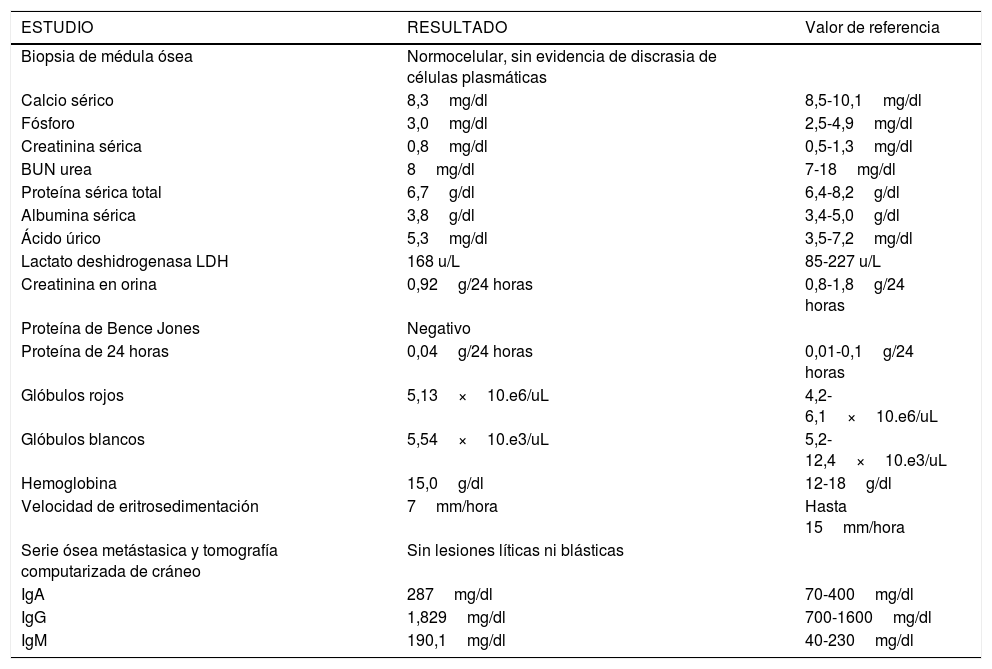
El paciente fue citado a consulta externa de hematoncológica donde se le realizaron estudios que descartaron la presencia de mieloma múltiple (tabla 1). Es importante mencionar que no se pudo realizar electroforesis de proteína, inmunofijación y cadenas ligeras libres en la sangre y orina por no contar con dichos estudios en el hospital. Posteriormente continuó seguimiento en consulta externa de oncología y un año después de la cirugía el paciente se encuentra en buen estado general sin sintomatología.
Pruebas Diagnósticas Complementarias
| ESTUDIO | RESULTADO | Valor de referencia |
|---|---|---|
| Biopsia de médula ósea | Normocelular, sin evidencia de discrasia de células plasmáticas | |
| Calcio sérico | 8,3mg/dl | 8,5-10,1mg/dl |
| Fósforo | 3,0mg/dl | 2,5-4,9mg/dl |
| Creatinina sérica | 0,8mg/dl | 0,5-1,3mg/dl |
| BUN urea | 8mg/dl | 7-18mg/dl |
| Proteína sérica total | 6,7g/dl | 6,4-8,2g/dl |
| Albumina sérica | 3,8g/dl | 3,4-5,0g/dl |
| Ácido úrico | 5,3mg/dl | 3,5-7,2mg/dl |
| Lactato deshidrogenasa LDH | 168 u/L | 85-227 u/L |
| Creatinina en orina | 0,92g/24 horas | 0,8-1,8g/24 horas |
| Proteína de Bence Jones | Negativo | |
| Proteína de 24 horas | 0,04g/24 horas | 0,01-0,1g/24 horas |
| Glóbulos rojos | 5,13×10.e6/uL | 4,2-6,1×10.e6/uL |
| Glóbulos blancos | 5,54×10.e3/uL | 5,2-12,4×10.e3/uL |
| Hemoglobina | 15,0g/dl | 12-18g/dl |
| Velocidad de eritrosedimentación | 7mm/hora | Hasta 15mm/hora |
| Serie ósea metástasica y tomografía computarizada de cráneo | Sin lesiones líticas ni blásticas | |
| IgA | 287mg/dl | 70-400mg/dl |
| IgG | 1,829mg/dl | 700-1600mg/dl |
| IgM | 190,1mg/dl | 40-230mg/dl |
Los plasmocitomas intracraneales son generalmente sintomáticos, la manifestación más común es el dolor resultado de la erosión ósea, los otros síntomas producidos dependen de la localización de la lesión, y así se han descrito: cefalea, crisis convulsivas, signos neurológicos focales, datos de hipertensión intracraneal, parálisis de nervios craneales para los que afectan la base, los que afectan a la duramadre frontal producen cambios de la personalidad y alteraciones cognoscitivas similares a las observadas en pacientes con lesión significativa del lóbulo frontal5.
En el caso de este paciente, no se presentó dolor como principal síntoma, ya que no hubo afectación ósea; sin embargo, por su ubicación en la hoz del cerebro y sumando la presencia de edema en la cisura medial que se extendió hasta el lóbulo frontal, presentó: diplopía, cefalea y prosopagnosia de lenta evolución pero que se exacerbaron acompañándose de signos de hipertensión endocraneana, ante este cuadro, se realizó tomografía computarizada que hizo sospechar meningioma.
El diagnóstico definitivo de plasmocitoma es histológico y no se considera como principal método diagnóstico radiológico ya que puede confundirse con otros tumores, especialmente el meningioma, puesto que en la tomografía computarizada ambas son lesiones extraaxiales iso o hiperdensos3,5,6,8. En este caso, el paciente fue ingresado al quirófano con sospecha de meningioma por lo que se realizó exceresis tumoral Simpson I, diagnóstico que fue sustituido con la identificación de la discrasia de células plasmáticas en el estudio histopatológico.
La resección quirúrgica anatómica adecuada y completa seguida de radioterapia es el tratamiento de elección, ya que el tumor es muy sensible a la radiación3,8. Inicialmente el paciente fue tratado con manitol, el cual resolvió los signos de hipertensión endocraneana, después se realizó exeresis tumoral, con resolución de síntomas. Antes de iniciar la radioterapia es necesario confirmar el diagnóstico histopatológico de plasmocitoma y descartar el desarrollo de mieloma múltiple, ya que este último precisa de quimioterapia. Los plasmocitomas extramedulares pueden ser los precursores de mieloma múltiple en entre 20 y 30% de los pacientes, este se puede desarrollar hasta varios años después del inicio de la enfermedad, y debe descartarse para decidir tratamiento5,8.
La Sociedad Americana del Cáncer y la Sociedad Española de Oncología Médica establecen los criterios diagnósticos del mieloma múltiple. La primera afirma que muchas personas con un plasmocitoma solitario padecerán mieloma múltiple, a estos pacientes se les observa cuidadosamente para identificar signos de esta enfermedad9,10,12. El criterio más importante para determinar que se trata de mieloma múltiple sintomático son las manifestaciones de daño a órgano blanco, que incluyen: anemia, hipercalcemia, insuficiencia renal, hiperviscosidad, amiloidosis, daño óseo o infecciones recurrentes11. En este caso se realizaron una serie de pruebas diagnósticas, las cuales descartaron la presencia actual de mieloma múltiple (tabla 1); sin embargo, es necesario un seguimiento a largo plazo aproximadamente de 10 a 12 años ya que la recurrencia o progresión a un mieloma múltiple ocurre del 10 al 30% de los casos4,12. Cabe mencionar que no se pudo realizar electroforesis de proteína, inmunofijación y las cadenas ligeras libres; no obstante, tanto la biopsia de medula ósea como la proteína de Bence Jones y las inmunoglobulinas se reportaron normales, no se encontraron lesiones osteolíticas, hematológicas, renales, hepáticas y no presentó artralgias u otra evidencia clínica que hiciera sospechar la presencia del mieloma múltiple.
El pronóstico para las formas aisladas es más benigno que el de un mieloma múltiple, por lo que descartar la presencia de este es importante ya que la mortalidad se debe a infecciones recurrentes y a insuficiencia renal con supervivencia de 2 a 3 años. La historia natural del mieloma múltiple es de deterioro progresivo con daño a diferentes órganos, lo que finalmente conduce a la muerte; sin embargo, la probabilidad de que un plasmocitoma extramedular progrese a un mieloma múltiple es menor a 30% con un período libre de enfermedad a los 10 años de 70%12,13.
El diagnóstico definitivo de las neoplasias de células plasmáticas requiere la combinación de los hallazgos histopatológicos con los hallazgos clínicos y de laboratorio14. Una vez descartada la presencia de mieloma múltiple se realizó confirmación de plasmocitoma por estudios de inmunohistoquímica, previamente la tinción de hematoxilina eosina reveló la presencia de células plasmáticas maduras que se caracterizan por presencia de núcleos excéntricos8. Las neoplasias de células plasmáticas expresan CD138 y CD79a pero son CD20 negativas, la expresión de este último se pierde en el proceso de maduración terminal de la célula B14, tal como se evidenció en este caso. CD45 resultó positivo, este constituye un conjunto de proteínas tirosinfosfatasa que se expresan prácticamente en todas las células hematolinfoides y sus precursores; con respecto al antígeno epitelial de membrana (EMA), este fue negativo ya que su función es para identificar tumores de origen epitelial, pueden ser positivos en algunos casos de: linfoma B rico en T, linfoma B difuso de células grandes y leucemia megacariocítica15.
Una neoplasia hematopoyética muestra constantes y consistentes defectos moleculares y expresa un grupo de antígenos constantes, la demostración de restricción en las cadenas ligeras kappa y lambda son buen indicador de monotipicidad en un proceso B15. En este caso se está ante un plasmocitoma secretor de cadenas ligeras lambda ya que fue positivo para este y negativo para kappa. Finalmente, Ki-67 es un antígeno nuclear presente en todas las células proliferantes que se encuentran en la fase activa del ciclo celular (G1, S, G2 y mitosis) y ausente en G0, y que puede ayudar a decantar el diagnóstico a favor de benignidad o malignidad en algunos casos, en este paciente se presentó un índice de proliferación bajo14.
El principal diagnóstico diferencial del plasmocitoma extramedular es el linfoma B de la zona marginal del tejido linfoide asociado a mucosas (MALT), el cual tiene una gran diferenciación plasmocelular y a veces muy pocos linfocitos B neoplásicos lo que conduce a esta confusión. Esto se afirma en la clasificación de la OMS del 2008, señalando que es posible que muchos casos diagnosticados como plasmocitomas extramedulares sean linfomas marginales B con marcada diferenciación plasmocelular dado su comportamiento indolente. Puesto que hay un 30% de linfomas del tejido linfoide asociado a mucosas (MALT) con diferenciación plasmocítica indistinguible histológicamente de un plasmocitoma, sin embargo, el estudio anatomopatológico e inmunohistoquímico permiten diferenciar el diagnóstico entre ambas16–18.
Debido a las similitudes, tanto en la clínica como en la neuroimagen que pueden compartir los meningiomas y plasmocitomas, es necesario una adecuada correlación con el diagnóstico histopatológico e inmunohistoquímico que permita al clínico investigar la presencia de mieoloma múltiple, ya que el plasmocitoma puede ser un indicador y comprometer la vida del paciente. También para la toma de decisiones terapéuticas, pues tanto el pronóstico como el uso de radioterapia o quimioterapia dependerán del diagnóstico diferencial entre meningioma y plasmocitoma.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
AgradecimientosAl doctor Danilo Alvarado por su ayuda para la realización de los estudios de inmunohistoquímica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
