



La enfermedad de Creutzfeldt-Jakob (ECJ) es un raro trastorno neurodegenerativo que se presenta con un rápido y progresivo deterioro de la memoria, cambios de comportamiento, falta de coordinación y/o alteraciones visuales. Esta forma parte de un grupo de enfermedades conocidas como encefalopatías espongiformes transmisibles (EET) o enfermedades priónicas, que son causadas por la acumulación de la proteína priónica patológica (PrP) en el sistema nervioso central, y que se puede producir de forma esporádica, hereditaria o adquirida. Los síntomas suelen comenzar alrededor de 60-70 años y a medida que avanza la enfermedad el daño neuronal progresivo debido a los depósitos de PrP conduce a un deterioro mental más pronunciado, a la aparición de trastornos del movimiento, ceguera y coma, alcanzando una tasa de mortalidad alta (90%) dentro del primer año.
La enfermedad de Creutzfeldt-Jakob (ECJ) es un raro trastorno neurodegenerativo que se presenta con un rápido y progresivo deterioro de la memoria, cambios de comportamiento, falta de coordinación y/o alteraciones visuales. Esta forma parte de un grupo de enfermedades conocidas como encefalopatías espongiformes transmisibles (EET) o enfermedades priónicas, que son causadas por la acumulación de la proteína priónica patológica (PrP) en el sistema nervioso central, y que se puede producir de forma esporádica, hereditaria o adquirida. Los síntomas suelen comenzar alrededor de 60-70 años y a medida que avanza la enfermedad el daño neuronal progresivo debido a los depósitos de PrP conduce a un deterioro mental más pronunciado, a la aparición de trastornos del movimiento, ceguera y coma, alcanzando una tasa de mortalidad alta (90%) dentro del primer año.
Se presenta un caso de una mujer de 65 años de edad, que acude a urgencias por alteración del lenguaje progresivo y postura anormal en su brazo derecho, que había comenzado un mes antes. Después de detallados exámenes clínicos y neuropsicológicos se sospechó de la ECJ y se completó el estudio con exámenes complementarios tales como EEG, punción lumbar y resonancia magnética del cerebro. La paciente presentó deterioro cognitivo rápido y global, lo que le lleva a la muerte 2 meses después del ingreso. El estudio anatomopatológico posterior confirmó el diagnóstico de ECJ.
Se presenta un caso de una mujer de 65 años de edad, que acude a urgencias por alteración del lenguaje progresivo y postura anormal en su brazo derecho, que había comenzado un mes antes. Después de detallados exámenes clínicos y neuropsicológicos se sospechó de la ECJ y se completó el estudio con exámenes complementarios tales como EEG, punción lumbar y resonancia magnética del cerebro. La paciente presentó deterioro cognitivo rápido y global, lo que le lleva a la muerte 2 meses después del ingreso. El estudio anatomopatológico posterior confirmó el diagnóstico de ECJ.
Es importante conocer la enfermedad para poder proporcionar una atención de enfermería adecuada. La rápida progresión de esta enfermedad y el nivel de dependencia hace necesario reevaluar continuamente este tipo de pacientes.
Es importante conocer la enfermedad para poder proporcionar una atención de enfermería adecuada. La rápida progresión de esta enfermedad y el nivel de dependencia hace necesario reevaluar continuamente este tipo de pacientes.
Creutzfeldt-Jakob disease (CJD) is a rare neurodegenerative disorder presenting with rapidly progressive memory impairment, behavioral changes, lack of coordination and/or visual disturbances. This condition is part of a group of diseases known as spongiform encephalopathies or prionic diseases, which are caused by accumulation of a pathological prion protein (PrP) in the central nervous system, and may occur in sporadic, hereditary or acquired form. Symptoms usually begin around 60-70 years old and, as the disease advances, progressive neuronal damage due to PrP deposits leads to a more pronounced mental deterioration, appearance of movement disorders, blindness and coma, reaching a high mortality rate (90%) within first year.
We present a case of a 65 year-old woman who came to the emergency room complaining of progressive language disturbance and abnormal posture of her right arm that had begun one month earlier. After detailed clinical and neuropsychological examinations CJD was suspected and complementary examinations such as EEG, brain MRI and lumbar puncture were performed. The patient showed rapid and global cognitive deterioration, leading to her death two months after admission. A subsequent pathological study confirmed the diagnosis of CJD.
It is important to know the disease in order to provide adequate nursing care. The rapid progression of the disease and the level of dependence make it necessary to continually reappraise these patients.
AbstractCreutzfeldt-Jakob disease (CJD) is a rare neurodegenerative disorder presenting with rapidly progressive memory impairment, behavioral changes, lack of coordination and/or visual disturbances. This condition is part of a group of diseases known as spongiform encephalopathies or prionic diseases, which are caused by accumulation of a pathological prion protein (PrP) in the central nervous system, and may occur in sporadic, hereditary or acquired form. Symptoms usually begin around 60-70 years old and, as the disease advances, progressive neuronal damage due to PrP deposits leads to a more pronounced mental deterioration, appearance of movement disorders, blindness and coma, reaching a high mortality rate (90%) within first year.
We present a case of a 65 year-old woman who came to the emergency room complaining of progressive language disturbance and abnormal posture of her right arm that had begun one month earlier. After detailed clinical and neuropsychological examinations CJD was suspected and complementary examinations such as EEG, brain MRI and lumbar puncture were performed. The patient showed rapid and global cognitive deterioration, leading to her death two months after admission. A subsequent pathological study confirmed the diagnosis of CJD.
It is important to know the disease in order to provide adequate nursing care. The rapid progression of the disease and the level of dependence make it necessary to continually reappraise these patients.
Aunque es posible que esta enfermedad se conociera desde la más remota antigüedad, sus síntomas inespecíficos deben haber sido confundidos con otros tipos de demencia durante siglos. Esta enfermedad fue descrita por primera vez por los neurólogos alemanes Hans-Gerhard Creutzfeldt y Alfons Maria Jakob en el año 1920.
La enfermedad de Creutzfeldt-Jakob (ECJ) es un trastorno poco frecuente (prevalencia 1:106) neurodegenerativo del cerebro y mortal. Por lo general, los síntomas comienzan a la edad de 60-70 años. Hay 3 categorías principales de la ECJ: esporádica, hereditaria y adquirida1.
Esta enfermedad está producida por una proteína llamada prión (PrP). Esta es en sí una proteína inocua presente en todas la células de nuestro cuerpo, y que está destinada a controlar ciertos aspectos de la vida celular. Cuando esta proteína adquiere una configuración diferente a la normal (plegada) se comporta de forma patológica. Es por este motivo que el sistema inmune no es capaz de luchar contra este prión, ya que en sí se trata de una proteína propia y se considera infecciosa, ya que una sola de estas proteínas es capaz de cambiar la configuración normal de sus similares.
En la ECJ adquirida el prión ingresa en el organismo a través del contacto con priones infecciosos. En la ECJ hereditaria el gen responsable de producir esta proteína (Prion-Related Protein [PRNP]) ha sufrido una mutación, por lo que solo es capaz de producir la proteína patológica.
Para conocer el modus operandi de estas proteínas hace falta describir el funcionamiento normal de las células nerviosas. Estas células poseen un receptor químico que normalmente está obstruido por un PrP (PrPc) normal. Debido a que este receptor no puede ser ocupado por el PrP patológico es cuando ciertas proteínas encargadas de destruir las neuronas enfermas o anormales introducen parte de su estructura en estos receptores, produciendo la destrucción celular.
El diagnóstico de esta enfermedad es muy difícil, ya que presenta sintomatología fácilmente atribuible a otras enfermedades neurodegenerativas.
El único método de diagnóstico es el estudio anatomopatológico del cerebro2.
Caso clínicoPaciente mujer de 65 años que consulta en Urgencias por alteración del lenguaje y postura distónica de la extremidad superior derecha.
Antecedentes personales- Hipertensión.
- Dislipidemia.
- Hipotiroidismo en tratamiento sustitutivo.
- No posee antecedentes familiares de enfermedades neurodegenerativas.
- No tiene antecedentes de haber residido en el Reino Unido.
- No ha sido sometida a ninguna transfusión.
Vive sola. Jubilada desde los 59 años. Directora de marketing en un laboratorio farmacéutico.
Valoración inicialEl mes anterior al ingreso la paciente presenta un cuadro de desorientación y alteración del lenguaje, por lo cual ingresa durante 6 días en otro hospital de nuestra zona de influencia. Se le realiza resonancia magnética nuclear (RMN), estudio vascular (eco doppler TSA y ecocardiograma), siendo diagnosticada de ictus isquémico agudo en la cabeza del núcleo del caudado izquierdo.
Consulta de nuevo en nuestro centro hospitalario por un aumento de la alteración del lenguaje y temblor en extremidad superior derecha.
Exploración neurológica al ingresoConsciente, desorientada en tiempo y espacio. Comprende órdenes simples, pero no complejas. Presenta alteración de la fluencia verbal. No presenta déficits motores en las extremidades. Temblor en la extremidad superior derecha. Dismetría dedo-nariz, talón-rodilla derecha. Deambula de forma autónoma, aunque presenta posición discretamente distónica.
Exploraciones complementariasRMN craneal: se observa una lesión isquémica en la cabeza del núcleo caudado izquierdo.
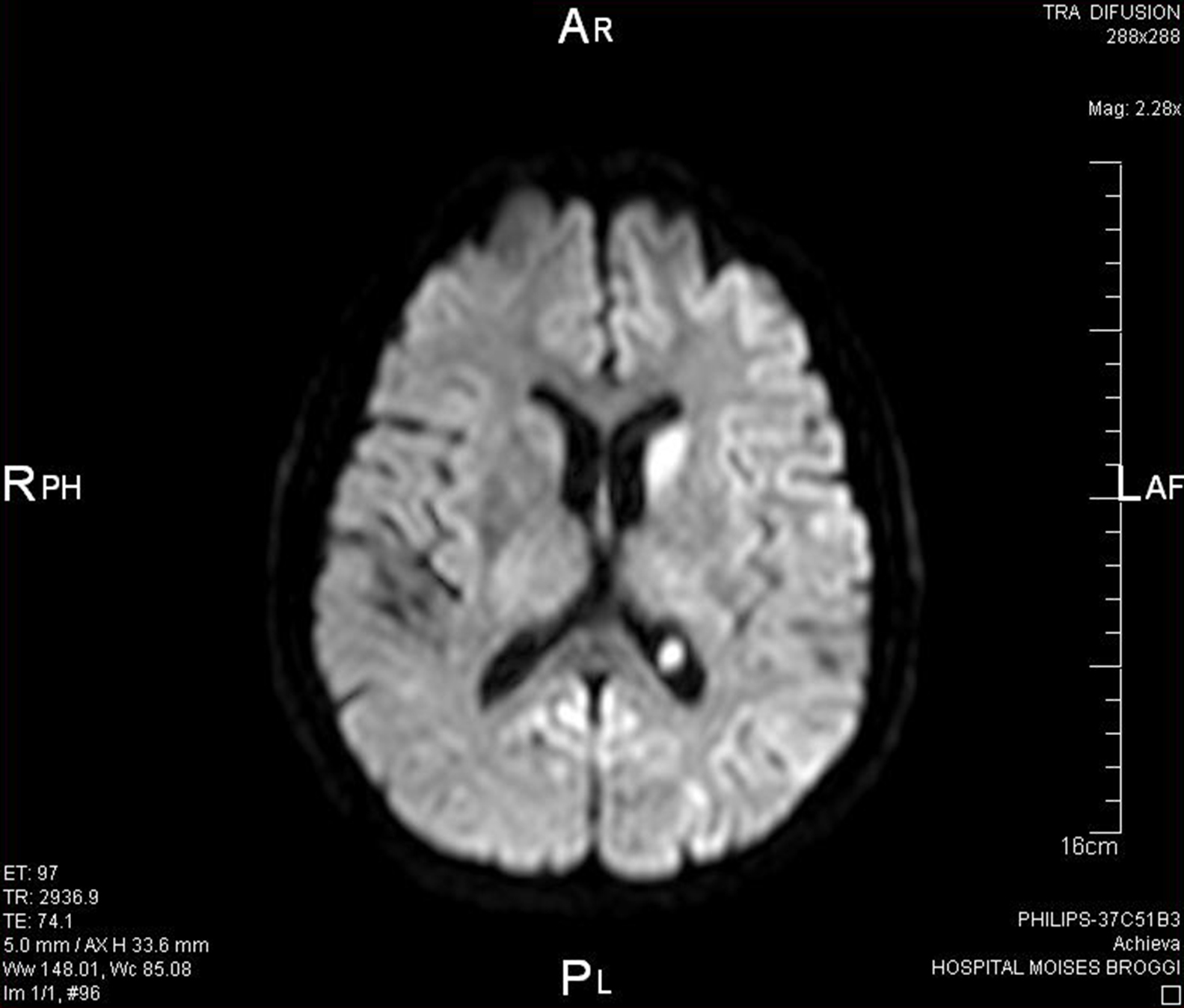
Al revisar de nuevo la RMN se observan más alteraciones de las que se pensaban en principio, con lo que el diagnóstico anterior de ictus isquémico es poco probable (Figura 1).
Figura 1. Resonancia magnética nuclear.
Se cursa analítica, punción lumbar (PL) con bioquímica, citología, inmunoglobulinas, bandas, serología, metales pesados y proteína 14-3-3.
Las serologías y el estudio inmunológico son negativos, la proteína 14-3-3 no se detecta en líquido cefalorraquídeo (LCR).
Dado que la paciente tiene hipotiroidismo con anticuerpos antitiroidales positivos se plantea como posible diagnóstico diferencial la encefalopatía de Hashimoto y se administra una pauta de 5 días de bolus de corticoides sin respuesta clínica.
El electroencefalograma (EEG) muestra disfunción global marcada de predominio anterior más acentuada en el hemisferio izquierdo, sugestiva a encefalopatía. Se realizan 3 EEG más de control, que arrojan el mismo resultado, y en el último presenta también actividad irritativa generalizada coincidente o no con clonías de baja amplitud, predominantemente cefálica y en el hemicuerpo derecho.
EvoluciónTras descartar otras posibles causas se empieza a sospechar como diagnóstico la ECJ.
En pocos días la paciente progresa de una alteración de la fluencia verbal a mostrar ya un discurso incoherente y prácticamente incomprensible, bloqueos del lenguaje constantes, junto con probables alucinaciones visuales, mano ajena derecha y confunde a los familiares. A nivel motor se empieza a observar mioclonías espontáneas en la extremidad superior derecha. Muestra marcha inestable por postura distónica.
A los 10 días de ingreso la paciente tiende a la somnolencia, solo articula monosílabos o frases cortas de forma automática. Presenta cuadros de desorientación y agitación nocturna.
A los 15 días está prácticamente mutista. La deambulación es muy dificultosa, incluso con apoyo bilateral. Presenta ataxia en extremidad superior derecha y temblor postural de gran amplitud, movimientos repetitivos en la mano derecha y dismetría en la extremidad inferior derecha. También se objetivan en algún momento sacudidas repetitivas de la cabeza en «no-no».
A los 20 días de ingreso van en aumento las mioclonías en todo el hemicuerpo derecho; sigue mutista.
A los 45 días la paciente está encamada, mutista y sin reaccionar a estímulos verbales. Presenta tetraparesia espástica y mioclonías espontáneas generalizadas y segmentarias. Negación y dificultad a la ingesta.
Casi a los 2 meses del ingreso la paciente se encuentra en estado terminal de la posible ECJ y prácticamente arreactiva a estímulos. Presenta mioclonías generalizadas, postura distónica predominante en el hemicuerpo derecho, sin apertura ocular espontánea, además de crisis comiciales frecuentes.
Fallece a los 2 meses del ingreso.
Se realiza necropsia que resulta positiva para la ECJ.
Plan de cuidados 3–5Tras realizar varias valoraciones de enfermería, utilizando el modelo de V. Hendenrson, identificamos los principales diagnósticos y elaboramos un plan de cuidados según la taxonomía NANDA-NIC-NOC (Tabla 1).
Tabla 1. Plan de cuidados según la taxonomía NANDA-NIC-NOC
| NANDA | NOC | NIC |
| 0014 Incontinencia fecal | 0500 Continencia intestinal 0501 Eliminación intestinal 1101 Integridad tisular y membranas mucosas | 0410 Cuidados de la incontinencia intestinal 3590 Vigilancia de la piel |
| 0015 Riesgo de estreñimiento | 0501 Eliminación intestinal | 0450 Manejo del estreñimiento |
| 0021 Incontinencia urinaria total | 0502 Continencia urinaria 0503 Eliminación urinaria 1101 Integridad tisular y membranas mucosas | 0610 Cuidados de la incontinencia urinaria 3590 Vigilancia de la piel |
| 0039 Riesgo de aspiración | 1010 Estado de deglución | 3200 Precauciones para evitar la aspiración |
| 0047 Riesgo de deterioro de la integridad cutánea | 1101 Integridad tisular y membranas mucosas | 3540 Prevención de úlceras por presión |
| 0051 Deterioro de la comunicación verbal | 0902 Comunicación | 4976 Mejorar la comunicación: déficit del habla |
| 0060 Interrupción de los procesos familiares | 2600 Afrontamiento de los problemas de la familia | 7040 Soporte al cuidador principal 7110 Fomento de la implicación familiar |
| 0085 Deterioro de la movilidad física | 0200 Ambular 0210 Realización trasferencia | 221 Terapia ejercicios: ambulación |
| 0102 Déficit de autocuidado: alimentación | 0300 Autocuidados: ABVD | 1803 Ayuda con los autocuidados: alimentación |
| 00108 Déficit de autocuidado: baño | 0300 Autocuidados: ABVD | 1801 Ayuda con los autocuidados: Baño/higiene |
| 00109 Déficit de autocuidado: vestido | 0300 Autocuidados: ABVD | 1802 Ayuda con los autocuidados: vestir y arreglo personal |
| 00110 Déficit de autocuidado: uso del inodoro | 0300 Autocuidados: ABVD | 1804 Ayuda con los autocuidados: Aseo, eliminación |
| 00173 Riesgo de confusión aguda | 0916 Nivel de confusión aguda 0901 Orientación cognitiva 004 Sueño | 1850 Mejorar el sueño 6440 Manejo del delirio |
| 00155 Riesgo de caídas | 1912 Caídas 1909 Conducta prevención de caídas | 6490 Prevención de caídas 6654 Vigilancia (seguridad) 6486 Manejo ambiental |
| 00146 Ansiedad | 1211 Nivel de ansiedad 00148 Temor | 4920 Escucha activa 5270 Soporte emocional |
| 00040 Riesgo de síndrome de desuso | 0204 Consecuencias de la inmovilidad fisiológicas 0205 Consecuencias de la inmovilidad psicocognitivas | 740 Cuidados al paciente encamado |
Se han identificado los principales diagnósticos de enfermería y elaborado un plan de cuidados.
Es necesario revalorar continuamente a este tipo de paciente dada la rápida evolución de este tipo de enfermedad y el gran nivel de dependencia que supone para el enfermo.
Hemos podido observar durante la realización de nuestro estudio que hay una estrecha relación entre la progresión de la enfermedad y los diagnósticos de enfermería detectados, aunque sería necesario complementar este estudio relacionando las diferentes valoraciones de enfermería con sus diagnósticos para poder dar validez a esta afirmación.
Conflicto de interesesLos autores de este artículo declaran que no existe conflicto de intereses alguno ni fuente de financiación.
Recibido 10 Febrero 2013
Aceptado 15 Abril 2013
Autor para correspondencia. chmunne@gmail.com