


Estimada Editora:
La enfermedad de Creutzfeldt–Jakob (ECJ) es una encefalopatía espongiforme que conduce a una demencia rápidamente progresiva, así como a otras alteraciones neurológicas no específicas. Se describen cuatro tipos1:
- •
Esporádica (ECJe): 85-90% de los casos. Tiene etiología desconocida.
- •
Variante (ECJv): descrita en 1996, se relaciona con la exposición a la proteína de la enfermedad de la vaca loca. Generalmente muestra un curso más largo que los otros tipos.
- •
Familiar (ECJf): 10% de los casos. Aparece en individuos portadores de la mutación de la proteína priónica (PrP).
- •
Iatrogénica: por contacto con tejido cerebral o líquido cefalorraquídeo (LCR) de pacientes infectados.
Presentamos el caso de una mujer de 72 años que acudió al servicio de Urgencias de nuestro hospital por tener alteraciones visuales de un mes de evolución, acompañadas, progresivamente, por alteraciones de la marcha, agrafia, alexia y bradilalia. Como antecedentes de importancia la paciente refería hipertensión, diabetes y dislipemia, y era independiente para las actividades de la vida diaria hasta el inicio del cuadro.
En el examen físico se objetivó un síndrome cerebeloso (dismetría dedo-nariz y talón-rodilla bilateral, disdiadocosinesia, Romberg positivo bilateral con los ojos cerrados, marcha atáxica con lateralización y aumento de base de sustentación y habla escandida), alteraciones visuales (visión en túnel binocular y alteración de la percepción de los colores), piramidalismo (espasticidad, reflejos osteotendinosos exaltados) y deterioro cognitivo (bradilalia, alexia y agrafia) de corta y rápida evolución. Los estudios de rutina y la tomografía cerebral fueron normales.
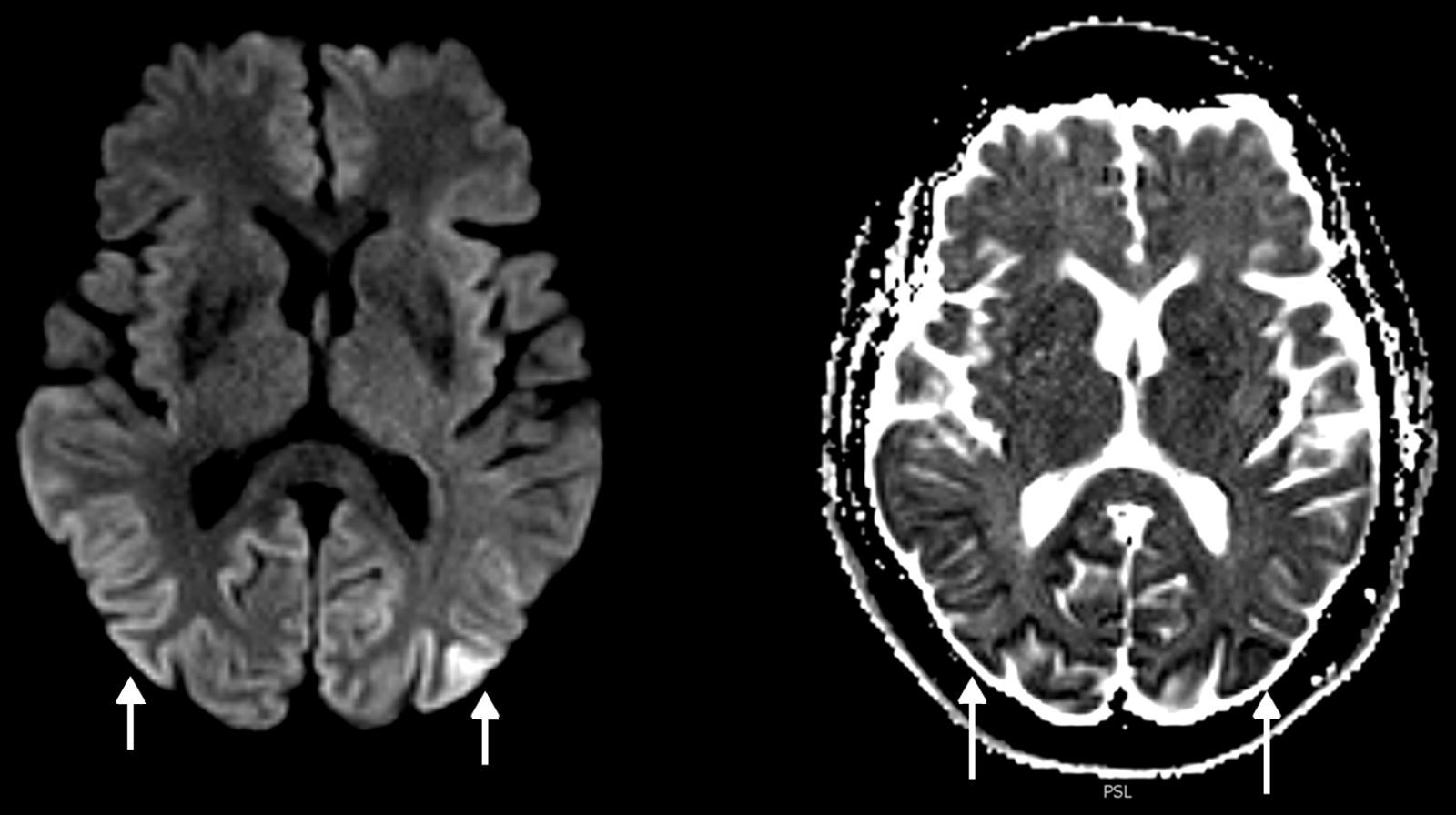
En una resonancia magnética (RM) cerebral hecha en un equipo de 3 Teslas (3T), que se había realizado 6 semanas antes en otro centro, se observaba restricción a la difusión en la corteza parieto-occipital bilateral. Estos hallazgos fueron confirmados y superpuestos a la RM (3T) realizada en nuestro centro al segundo día del ingreso (fig. 1). A los dos meses se llevó a cabo una RM de control con hallazgos similares, aunque menos evidentes, al ser hecha con un campo abierto de 1T.
: hiperintensidad de señal en la corteza parieto-occipital bilateral, que se muestra hipointensa en el mapa de ADC (flechas), confirmándose la restricción a la difusión.")
El electroencefalograma (EEG) evidenció signos de afectación cerebral generalizada de intensidad severa más acusados en el hemisferio derecho, con grafoelementos de tendencia pseudoperiódica.
El estudio en el LCR de la proteína 14-3-3 fue positivo, por lo que, unido a las manifestaciones clínicas y hallazgos de imagen, se estableció un diagnóstico probable de ECJe con variante de Heidenhain2.
La ECJ se caracteriza por demencia progresiva rápida, atrofia cerebral, mioclonías y finalmente la muerte. Los pacientes con ECJv presentan en su mayoría síntomas sensoriales y psiquiátricos, mientras que la ECJe suele mostrar deterioro cognitivo progresivo, cambios de personalidad, alucinaciones y síntomas cerebelosos. Por su parte, la variante de Heidenhain es una forma infrecuente de ECJe, descrita por primera vez en 1929. En ella, se destaca la presencia de distintas alteraciones visuales, lo que suele retrasar y dificultar su diagnóstico2,3.
La Organización Mundial de la Salud (OMS) ha desarrollado criterios diagnósticos, catalogando los casos como definitivos, probables o posibles, en base a la presencia de la proteína 14-3-3 en el LCR, cambios en el EEG y clínica. El EEG se caracteriza por descargas síncronas periódicas, más frecuentes en las etapas tardías de la enfermedad. Sin embargo, la proteína 14-3-3 en el LCR es marcador bioquímico importante en la ECJ, su presencia no es patognomónica y requiere confirmación histopatológica.
Se trata, por tanto, de una enfermedad de difícil diagnóstico, especialmente en los estadios iniciales, que se puede confundir con otras causas de demencia4. Si bien en la actualidad la RM no se encuentra entre los criterios diagnósticos de la OMS, en la práctica clínica contribuye de manera muy significativa a la orientación diagnóstica5,6.
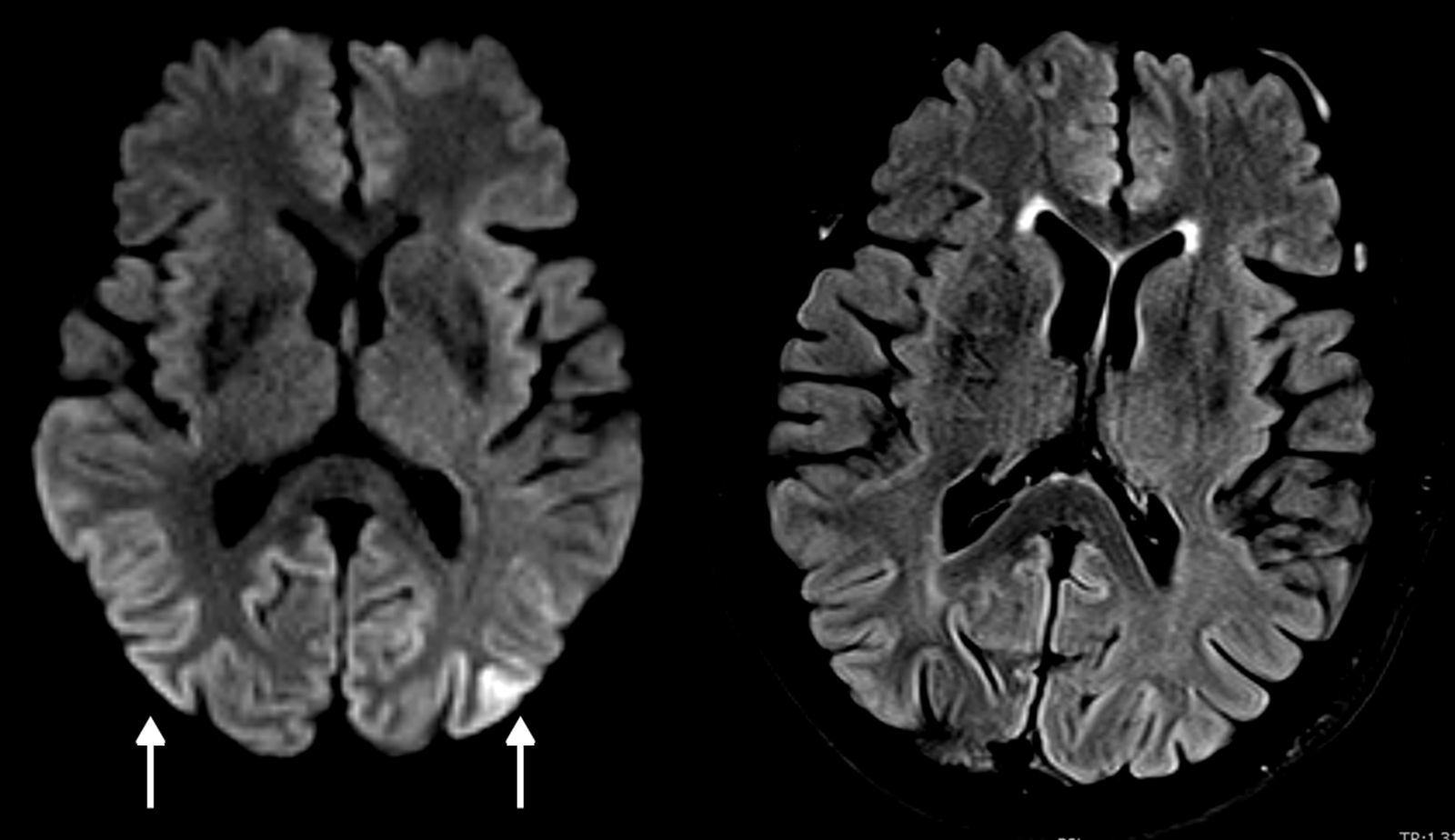
Los hallazgos de la RM puede ser uni o bilaterales y simétricos o asimétricos, e incluyen hiperintensidad en ponderación T2 y en atenuación del fluido en inversión-recuperación (FLAIR) de los ganglios basales (putamen y caudado), tálamo (signo del palo de hockey), corteza (manifestación temprana más común) o sustancia blanca4,5,7,8. En la ECJv los cambios se observan típicamente en el tálamo (signo del pulvinar). En la secuencia de difusión (DWI) es característica la difusión restringida persistente, especialmente evidente en los equipos de alto campo, siendo esta la secuencia más sensible para las alteraciones corticales, además de ser superior a las secuencias ponderadas en T2 y FLAIR en estadios tempranos2,7 (fig. 2). En la variante de Heidenhain son características las alteraciones de señal de los ganglios basales y típicamente del cortex occipital2,3. También se produce atrofia cerebral progresiva rápida.
 en la secuencia DWI en equipo de 3T, mucho menos evidentes en la secuencia FLAIR. La secuencia de difusión es la más sensible para las alteraciones corticales.")
El patrón de hiperintensidad de señal en FLAIR/restricción de la difusión puede llegar a diferenciar la ECJe de otras demencias rápidamente progresivas no priónicas, a pesar de que los hallazgos de imagen pueden superponerse8.
El diagnóstico diferencial debe hacerse con otras patologías que causen demencia. La enfermedad de Alzheimer no presenta alteraciones de la difusión en la RM y en las demencias vasculares la restricción no es persistente. Si la restricción a la difusión se limita al cortex cerebral, los principales diagnósticos diferenciales son el síndrome MELAS (miopatía, encefalopatía, acidosis láctica y episodios ictus like, que produce severo engrosamiento cortical hiperintenso en ponderación T2 y FLAIR), la encefalopatía posterior reversible y la encefalitis vírica, especialmente herpética (hinchazón cerebral típicamente temporal con cambios necrótico-hemorrágicos)4.
Aún no existe un tratamiento curativo y la enfermedad es invariablemente fatal, con una supervivencia media de siete meses tras el diagnóstico.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Conflicto de interesesLos autores del trabajo declaran no tener ningún conflicto de intereses.
