



La hipofisitis linfoplasmocitaria con expresión de inmunoglobulina G4 (IgG4) es una entidad de reciente conocimiento. Pertenece al grupo de enfermedades relacionadas a IgG4 (IgG4-RD, del inglés: IgG4-related disease), donde uno o varios órganos pueden estar comprometidos, con síntomas compresivos u obstructivos, o disfuncionalidad por infiltración celular. La hipófisis puede estar afectada en forma aislada. Clínicamente, se presentan con diabetes insípida, hipopituitarismo y/o síntomas de masa ocupante selar, siendo los principales diagnósticos diferenciales los adenomas selares no secretantes, y otros tipos de hipofisitis. Para arribar al diagnóstico de este tipo patología es necesaria la presencia de una imagen de agrandamiento selar o engrosamiento del tallo pituitario en la resonancia magnética nuclear, una histopatología característica con inmunomarcación positiva para IgG4 en más de 10 células plasmáticas por campo de gran aumento y la presencia de IgG4 sérica elevada. Tienen una excelente respuesta a glucocorticoides, por lo que una sospecha diagnóstica oportuna evitaría una cirugía innecesaria en la mayoría de los pacientes con esta entidad.
Immunoglobulin G4 (IgG4)-related lymphoplasmacytic hypophysitis is a recently known entity. It belongs to the IgG4-related diseases (IgG4-RD), in which one or more organs may be involved, with compressive or obstructive symptoms, or dysfunctionality due to cellular infiltration. The pituitary gland can be isolatedly affected. Clinically, lymphoplasmacytic hypophysitis presents with diabetes insipidus, hypopituitarism and/or symptoms of an occupying sellar mass, being the non-secreting sellar adenomas and other types of hypophysitis the main differential diagnosis. In order to reach the diagnosis, the presence of pituitary enlargement or pituitary stalk thickening on an MRI scan, a distinctive histopathology with positive for IgG4 immunostaining in more than 10 plasma cells per high-power field, and elevated serum IgG4 levels, confirms this type of hypophysitis. As this entity has an excellent response to glucocorticoids, the diagnosis suspicion may avoid an unnecessary surgery in most patients.
La hipofisitis es una entidad rara, causada por una inflamación crónica de la glándula hipófisis, que provoca diferentes grados de hipopituitarismo. Representa el 0,24 al 0,88% del total de patologías hipofisarias, con una incidencia anual de 1caso cada 9 millones de habitantes1,2. El diagnóstico es generalmente presuntivo, basado en la historia clínica y datos bioquímicos, y sustentado por una imagen selar en la RM.
La cirugía suele ser necesaria para arribar a un diagnóstico histopatológico y en el tratamiento de efectos compresivos de masa3.
Las hipofisitis linfoplasmocitarias con expresión de IgG4 pertenecen al grupo de hipofisitis primarias, que son aquellas que no tienen causa identificable, en contrapartida con las secundarias, que son las que forman parte de un cuadro sistémico conocido. Afectan principalmente a hombres en la séptima década de la vida y pueden presentarse con diabetes insípida, síntomas de masa ocupante selar o hipopituitarismo4. Pertenecen a las enfermedades relacionadas con la expresión de IgG4 (IgG4-RD), condición que compromete a múltiples órganos compartiendo una apariencia histológica común, con infiltrados linfocitarios policlonales y células plasmáticas con inmunomarcación positiva para IgG4, con progresión a la fibrosis y la disfuncionalidad, niveles séricos elevados de IgG4 y una respuesta rápida a la terapia inmunosupresora5,6.
El objetivo es presentar las principales características de las hipofisitis linfoplasmocitarias con expresión de IgG4, patología poco frecuente, descripta recientemente, cuya sospecha oportuna permitiría instaurar un tratamiento médico precoz y evitaría una cirugía hipofisaria innecesaria (en la mayoría de los casos).
HipofisitisLas hipofisitis pertenecen al grupo de masas selares no secretantes, compartiendo con ellas algunas características clínicas y radiológicas. Estas entidades comprenden un espectro clínico-patológico complejo, siendo las formas más frecuentes la linfocitaria y la granulomatosa, habiéndose descripto recientemente una nueva variante: la linfoplasmocitaria con expresión de IgG47,8.
Se las puede clasificar en primarias, cuando no tienen causa identificable, o secundarias, cuando se presentan en el contexto de una enfermedad sistémica de causa conocida. Las primarias se subclasifican según su histopatología en hipofisitis linfocitaria, granulomatosa, xantomatosa, necrosante y linfoplasmocitaria con expresión de IgG4. También se las puede clasificar según comprometan clínica o radiológicamente la hipófisis anterior, el lóbulo posterior y el tallo hipofisario, o ambos, en: adenohipofisitis, infundibuloneurohipofisitis o panhipofisitis, respectivamente. Las hipofisitis primarias pueden presentarse, a su vez, en forma aislada o como parte de enfermedades sistémicas, tales como los síndromes poliglandulares autoinmunes y las IgG4-RD7.
Las hipofisitis secundarias incluyen casos en los que se puede identificar claramente el agente causal, tal como la administración de tratamiento inmunomodulador como el interferón alfa o el ipilimumab, o casos en los que la hipófisis está comprometida dentro del contexto de una enfermedad sistémica, como la tuberculosis, enfermedad inflamatoria intestinal, granulomatosis de Wegener o histiocitosis, entre otras6,7.
La hipofisitis linfocitaria es la más común de las formas primarias. Es de origen autoinmune, con un gran predominio en mujeres respecto a hombres (relación 8:1), con edad promedio de 34 años. Si bien fue típicamente descripta durante el tercer trimestre de embarazo y los 2 primeros meses de puerperio, solo un 40% del total de casos ocurre en estos períodos vitales7, pudiendo afectar también a mujeres durante la menopausia9.
Le siguen en frecuencia las granulomatosas y las xantomatosas, que tienen un ligero predominio en mujeres, no relacionadas con el embarazo o el puerperio, y la necrosante, que afecta a jóvenes y niños. El diagnóstico de certeza de hipofisitis se logra tras el examen histopatológico del tejido biopsiado o resecado quirúrgicamente7.
Enfermedades relacionadas a IGG4Las IgG4-RD constituyen una entidad patológica emergente, de causa desconocida y con compromiso multiorgánico10. Son 3 veces más comunes en hombres de edad media o adultos mayores11. Es una condición fibroinflamatoria crónica caracterizada por lesiones con denso infiltrado linfoplasmocitario, rico en células plasmáticas con inmunomarcación positiva para IgG4, fibrosis estoriforme (fibroblastos dispuestos a modo de rayos de una rueda) y frecuente, aunque no constante, elevación de niveles séricos de IgG46.
Fueron descriptas por primera vez en el año 2001 en Japón, a partir del estudio de la pancreatitis fibrosante12. Varias enfermedades previamente descriptas como de origen idiopático, como la tiroiditis de Riedel, el síndrome de Miculicz, la fibrosis retroperitoneal, la nefritis túbulo-intersticial, entre otras, son actualmente consideradas parte del espectro de estas enfermedades, compartiendo características histopatológicas comunes independientemente del órgano afectado11.
Varios son los órganos que pueden estar involucrados, como páncreas, vía biliar, glándulas lagrimales, glándulas salivales, sistema nervioso central, tiroides, pulmón, tracto gastrointestinal, riñón, próstata, retroperitoneo, arterias, ganglios linfáticos, piel y mamas, en forma sincrónica o metacrónica. Las lesiones pueden ser nodulares o hiperplásicas6.
Tienen un curso clínico solapado, con manifestaciones que varían dependiendo de la localización, pudiendo algunos pacientes experimentar complicaciones serias, como síntomas compresivos u obstructivos debidos a organomegalia o hipertrofia; o disfunción por infiltración celular y fibrosis6.
En general, se admite que la presencia de marcación de IgG4 en > 10 plasmocitos por campo de gran aumento alcanza para hacer el diagnóstico, aunque este valor de corte puede variar según el órgano afectado y el tiempo de evolución de la fibrosis6.
La concentración de IgG4 sérica puede orientar, pero hasta en un 40% de los casos puede ser normal. Se utiliza preferentemente la relación IgG4/IgG total, que en general es mayor del 40%11.
Se caracterizan por presentar excelente respuesta inicial a glucocorticoides. El principal diagnóstico diferencial es con el linfoma de células B, que se descarta por la presencia de linfocitos B, mientras que en las IgG4-RD hay un infiltrado rico en linfocitos T. La presencia de granulomas, necrosis o neutrófilos nos aleja del diagnóstico de IgG4-RD10,12.
Hipofisitis linfoplasmocitarias con expresión de IgG4Es el subtipo de hipofisitis más recientemente descripto, siendo el primer caso reportado en el año 2004 sobre la base de datos clínicos13,14, y en el año 2007 con confirmación anatomo-patológica15.
EpidemiologíaEn la actualidad, hay descriptos un total de 43 casos de hipofisitis linfoplasmocitaria con expresión de IgG4, de los cuales solo 17 tienen confirmación por biopsia de hipófisis16-20. Lo que las diferencia del resto de las hipofisitis primarias es que es más frecuente en hombres, con una relación 3:1 respecto a mujeres, y son individuos de mayor edad, con una media de 68±8 años. Si bien la prevalencia ha sido difícil de calcular dado el limitado número de casos reportados, un estudio japonés ha establecido la misma a partir del análisis de 170 pacientes que consultaron a un hospital por hipopituitarismo y/o diabetes insípida, estimando que esta entidad tiene una frecuencia del 30% del total de hipofisitis, correspondiendo al 4% del total de causas de hipopituitarismo y diabetes insípida19.
De los casos publicados, el 77% afecta a población japonesa, así como el 30% del total de hipofisitis linfocitarias compromete a este grupo poblacional. Aún se desconoce si este fenómeno se debe a un mayor riesgo geográfico o étnico verdadero o si simplemente en Japón tienen más conocimiento de esta entidad en relación con otros países4.
PatogénesisLa patogénesis de las hipofisitis linfoplasmocitarias con expresión de IgG4, así como la de las IgG4-RD en general, está siendo foco de una intensa investigación. Los niveles séricos elevados de IgG4 y el infiltrado denso en células plasmáticas IgG4 positivas a la inmunomarcación en varios órganos indican que esta inmunoglobulina desempeña un rol central, aunque el gatillo para generar su elevación no ha sido claramente establecido21.
La IgG4 es la menos abundante de las inmunoglobulinas G (IgG) en personas sanas, correspondiendo a menos del 4% del total. Tiene características distintivas que la hacen única, tales como la incapacidad para activar el complemento, débil capacidad para la opsonización, escaso paso a través de la placenta, y muy baja afinidad de unión a receptores Fc expresados en mastocitos y basófilos.
Además, la presencia de puentes disulfuro que unen inestablemente las cadenas pesadas, permite su fácil separación conformando 2 sitios de unión antigénicos, por lo que el anticuerpo es biespecífico asimétrico, no pudiendo formar complejos inmunes. El significado de esta biespecificidad requiere mayor investigación, dado que sería relevante solo en situaciones donde se encuentren altos niveles de IgG4 asociados a 2 antígenos no relacionados, en el mismo tiempo y lugar12,16,17.
La producción de IgG4 está mediada por linfocitos T helper tipo 2 (LTh2). Las citocinas interleucinas (IL) 4 y 13 producidas por estos linfocitos aumentan la producción de IgG4 e inmunoglobulina E (IgE), mientras que la interleucina 10 (IL-10) favorece la producción preferencial de IgG4. Esta activación de los LTh2 con aumento concomitante de IgE y eosinófilos explicaría la mayor relación de las IgG4-RD y los antecedentes en algunos pacientes de asma y atopia. La principal función de la IgG4 sería la de interferir con la respuesta inmunitaria inducida por anticuerpos unidos al complemento, o respuestas mediadas por IgE. El origen alérgico podría explicar la mayor prevalencia de esta enfermedad en hombres de mayor edad y no en mujeres jóvenes, como ocurre con la hipofisitis linfocitaria4,12,22,23.
Se han reportado autoantígenos candidatos en la patogenia, como la hormona de crecimiento y la proopiomelanocortina, en pacientes con diagnóstico de esta entidad confirmado por biopsia, aunque más estudios deberían llevarse a cabo antes de generalizar y determinar que estos son verdaderos autoantígenos patogénicos en este u otro tipo de hipofisitis24,25.
Presentación clínicaClínicamente, la forma más común de presentación es con poliuria y polidipsia por diabetes insípida relacionada con la falta de producción de hormona antidiurética (58% de los casos), seguido por síntomas asociados a compromiso adrenocortical por déficit de adrenocorticotrofina, tales como malestar, fatiga, disminución del apetito, pérdida de peso, náuseas y vómitos (42% de los casos). Finalmente, los pacientes suelen presentar síntomas compresivos por masa ocupante selar, con trastornos visuales y cefaleas (15% de los casos). Al igual que en el resto de las hipofisitis, el compromiso de hipófisis posterior con diabetes insípida y del eje adrenocortical es un rasgo que las distingue de los adenomas hipofisarios, donde el eje adrenocortical es el más conservado13,26.
La mayoría de los 43 casos publicados presentó manifestaciones relacionadas con hipofisitis luego de la remisión de alguna IgG4-RD. Algunos presentaron manifestaciones de hipofunción pituitaria concomitantemente a la enfermedad sistémica y más raramente la hipofisitis antecedió al compromiso de otro órgano. Solo 9 pacientes se presentaron con hipofisitis como única expresión de la enfermedad. Las localizaciones más frecuentemente asociadas fueron: páncreas, pulmón, retroperitoneo y glándulas submandibulares, y con menos frecuencia: glándulas lagrimales, ganglios linfáticos, riñón, tiroides, órbita y paquimeninges27,28.
Por lo expuesto, los pacientes con IgG4-RD deberían tener un seguimiento cercano para evaluar el compromiso pituitario, principalmente diabetes insípida e hipocortisolismo, así como también los pacientes con este tipo de hipofisitis deberían ser estudiados para descartar compromiso de otros órganos19.
Es importante recordar que se debe realizar siempre una correcta anamnesis y examen clínico completo, de manera de descartar diagnósticos diferenciales tales como linfoma, germinoma, sarcoidosis, tuberculosis y neoplasias que pudieran ser el origen del compromiso pituitario.
El diagnóstico diferencial debe hacerse con las masas ocupantes selares no secretantes, entre ellas los adenomas y otros subtipos de hipofisitis. Se distingue de los adenomas en que estos excepcionalmente se presentan con diabetes insípida y tienen características típicas en la RM.
Estudios complementariosAdemás de los estudios hormonales, que son útiles para evaluar la integridad de los ejes hipotálamo-hipofisarios, contamos con la RM y los dosajes de IgG total e IgG4 séricas para arribar al diagnóstico, así como para el seguimiento y la evaluación de la respuesta terapéutica.
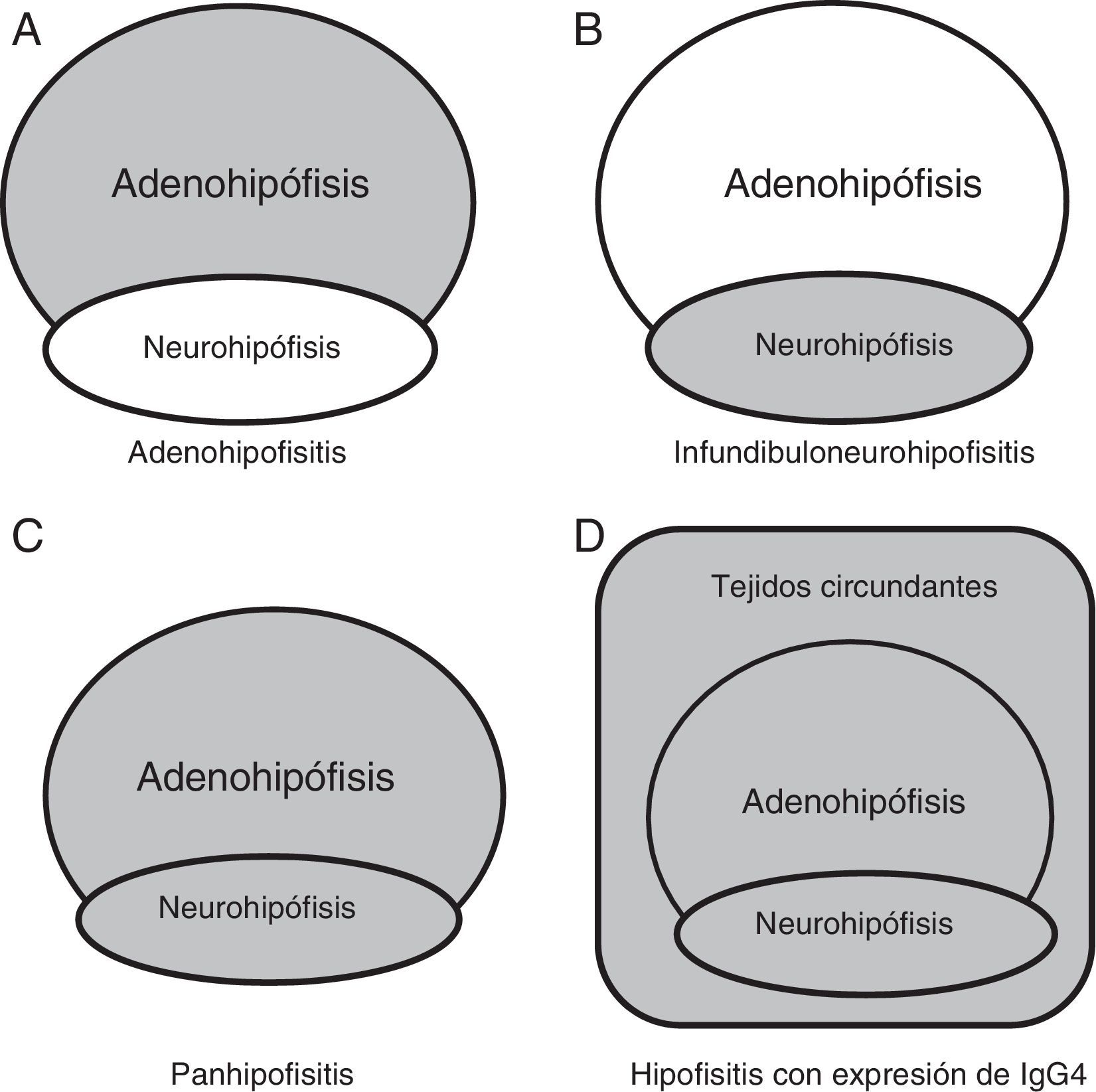
Resonancia magnética nuclearLa RM es el método de imagen de elección. Igual que para las hipofisitis linfocitarias, nos permite evidenciar un agrandamiento de la hipófisis anterior y/o engrosamiento del tallo hipofisario, con tinción homogénea de la glándula tras la inyección de gadolinio, frecuentemente asociada a la pérdida del brillo fisiológico de hipófisis posterior en secuencias T1 sin contraste4. En varios casos se ha encontrado asociación con paquimeningitis hipertrófica, seudotumor orbitario y parasinusitis, indicando que el proceso inflamatorio involucra no solo estructuras selares, sino también paraselares, rasgo que las distingue del resto de las hipofisitis (fig. 1)21. Algunos pacientes pueden presentar imágenes atípicas, con irregularidad de los bordes, heterogeneidad, destrucción de la silla turca e invasividad del seno esfenoidal8,18.
 Hipófisis anterior en adenohipofisitis. B) Hipófisis posterior con/sin afectación de tallo hipofisario en infundibuloneurohipofisitis. C) Ambas regiones en panhipofisitis. D) Compromiso selar y paraselar en las hipofisitis linfoplasmocitarias con expresión de IgG4. Adaptado de Shimatsu et al.21.")
Compromiso anatómico. A) Hipófisis anterior en adenohipofisitis. B) Hipófisis posterior con/sin afectación de tallo hipofisario en infundibuloneurohipofisitis. C) Ambas regiones en panhipofisitis. D) Compromiso selar y paraselar en las hipofisitis linfoplasmocitarias con expresión de IgG4.
Adaptado de Shimatsu et al.21.
Los niveles séricos de IgG4 están típicamente elevados, pudiendo alcanzar valores de hasta 10 veces el normal. Sin embargo, hasta en un 30% de los pacientes se encuentra dentro de la normalidad, a pesar de presentar hallazgos histopatológicos e inmunohistoquímicos típicos. Sus niveles disminuyen tras el inicio del tratamiento con glucocorticoides y en estadios tardíos de la enfermedad. El porcentaje de descenso y el tiempo que se requiere para alcanzar la normalidad son muy variables8,17.
HistopatologíaHistológicamente se presentan como un infiltrado inflamatorio linfoplasmocitario con grados variables de fibrosis, rico en células plasmáticas con inmunomarcación positiva para IgG4. El valor de corte es 10 o más células plasmáticas IgG4 positivas por campo de gran aumento, lo cual constituye generalmente más del 50% del total de IgG. Esto sumado a la positividad para cadenas livianas kappa y lambda revelan la presencia de un infiltrado policlonal4,14. La fibrosis es de tipo estoriforme, rasgo histológico característico de las IgG4-RD. Otro rasgo típico de estas enfermedades sistémicas es la flebitis obliterante, que no ha sido observada en biopsias de tejido hipofisario en los casos publicados hasta el momento19.
Criterios diagnósticoExisten 5 criterios diagnóstico propuestos por Leporati et al. en el año 20118: el primero es la presencia de un infiltrado mononuclear rico en linfocitos y células plasmáticas en la biopsia de hipófisis, con expresión de IgG4 en más de 10 células plasmáticas por campo de gran aumento; el segundo es la evidencia en imágenes de una masa selar o engrosamiento del tallo hipofisario; el tercero es la demostración por biopsia en otro órgano de inmunomarcación positiva para IgG4; el cuarto es el aumento de los niveles séricos de IgG4 (> 140mg/dl), y el quinto es la disminución del tamaño de la lesión y/o la mejoría de los síntomas (cefaleas y/o alteraciones visuales) con el tratamiento con corticoides. El diagnóstico se puede hacer de 3 maneras: solo con el primer criterio (criterio 1); con imágenes de masa selar en un paciente con confirmación histológica de inmunomarcación positiva para IgG4 en otro órgano (criterios 2+3), o con una imagen de masa selar en un paciente con niveles séricos de IgG4 elevados, que responde a tratamiento con corticoides (criterios 2+4+5) (tabla 1).
Criterios diagnóstico de hipofisitis linfoplasmocitarias con expresión de IgG4. Adaptado de Leporati et al.8

El criterio 1 de Leporati et al. está disponible claramente en una minoría de casos, lo cual es lo apropiado, ya que la cirugía pituitaria debería reservarse para casos excepcionales, teniendo en cuenta que la enfermedad responde muy bien a tratamiento médico8.
TratamientoEn relación con el tratamiento, cuanto antes se instale la terapia inmunosupresora, mayores serán las chances de conservación de la función pituitaria y la recuperación posterior29.
Los glucocorticoides están recomendados como de primera línea. Generalmente, se indica una dosis de prednisolona de 0,6mg/kg/día durante 2 a 4 semanas, disminuyendo la dosis de a 5mg cada 1 a 2 semanas por 2 a 3 meses para lograr la dosis de mantenimiento (2,5-5mg/día), la cual debería discontinuarse dentro de los 3 años de tratamiento29.
También se han reportado casos con respuesta favorable tanto clínica como imagenológica a dosis fisiológicas de reemplazo con hidrocortisona24,30.
Tal como ocurre en otras hipofisitis primarias, algunos pacientes presentan recaídas tras el descenso o la suspensión de glucocorticoides, requiriendo la instauración de otras medidas terapéuticas, como la azatioprina u otros inmunomoduladores, o la cirugía transesfenoidal15,25.
ConclusionesLas hipofisitis linfoplasmocitarias con expresión de IgG4 constituyen una entidad recientemente descripta, de causa desconocida, con características clínicas e histopatológicas distintivas. Si bien es una enfermedad rara, su prevalencia ha sido subestimada y es cada vez más reconocida por médicos clínicos y endocrinólogos.
Debemos sospecharla ante un paciente con diabetes insípida, hipopituitarismo o síntomas de masa ocupante selar. La hipófisis puede estar comprometida en forma aislada, o presentarse en el cuadro de IgG4-RD, es por esto que siempre hay que buscar compromiso de otros órganos en pacientes con afección pituitaria, así como buscar signos/síntomas de afección pituitaria en pacientes con IgG4-RD. Para esto se aconseja un manejo multidisciplinario del paciente.
La sospecha diagnóstica precoz permitirá la pronta instauración de un tratamiento médico con glucocorticoides y evitará una cirugía pituitaria innecesaria. El curso clínico de esta nueva entidad es difícil de predecir y tampoco se conoce la efectividad de la respuesta clínica de los glucocorticoides a largo plazo, por lo que se recomienda un seguimiento cercano y en el tiempo.
Finalmente, resta mucho por conocer en cuanto a la etiopatogenia de este tipo de hipofisitis, así como de las IgG4-RD en general. Un origen alérgico ha sido propuesto, por lo que en un futuro nuevas terapias podrían surgir con el objetivo de bloquear el agente gatillo de esta respuesta inflamatoria sistémica.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no poseer conflictos de interés.
Referencias no citadasA la Dra. Patricia Fainstein Day, por su mirada crítica y apoyo incondicional que hicieron posible la realización de esta monografía, y a todos los médicos del Servicio de Endocrinología, Metabolismo y Medicina Nuclear del Hospital Italiano de Buenos Aires, por el aporte académico invalorable en mi formación y por su calidad humana.