

INTRODUCCIÓN
La calcificación y osificación extraesqueléticas son patologías que observamos frecuentemente en la práctica clínica. Sin embargo, existen pocos informes en la literatura médica y el conocimiento acerca de estas entidades y su tratamiento es aún muy precario.
Guy Patin1, en 1962, describió el caso de una mujer que adquirió una consistencia dura en algunos tejidos, comparable a la de la madera (dure comme du bois). Munchmeyer2, en 1869, denominó a esta enfermedad miositis osificante progresiva. Teissier3, en 1876, describió unos depósitos calcáreos en una mujer de 21 años en diferentes sitios del cuerpo y en 1878 Weber4, los asocia por primera vez a la esclerodermia.
El depósito de calcio y fosfatos a nivel extraesquelético se presenta frecuentemente por la precipitación de sales amorfas, las cuales constituyen conglomerados de cristales de hidroxiapatita. En otros casos, en cambio, se evidencia una verdadera neoformación ósea5,6. Los términos «calcificación distrófica» y «calcinosis» se han utilizado para designar diferentes tipos de depósitos cálcicos, pero realmente el significado es el mismo: depósitos de calcio en tejidos extraesqueléticos, como el tejido subcutáneo, tejido desvitalizado o previamente lesionado5,6. El mecanismo de calcificación a nivel de los diferentes tejidos biológicos ha sido objeto de muchas investigaciones. Se han podido identificar depósitos de calcio en plantas así como en animales vertebrados e invertebrados, siempre en asociación con una matriz orgánica5,6. La mayoría de los depósitos patológicos de calcio suelen presentarse como cristales de hidroxiapatita, pero generalmente se carece de la estructura de la matriz osteoide en el sitio del depósito. No existe evidencia clara acerca de los fenómenos metabólicos que rigen los depósitos de calcio. Para que se presente el depósito de calcio aberrante en los diferentes tejidos, se requiere una alteración estructural en el área comprometida como ocurre en la calcinosis de la esclerosis sistémica progresiva (ESP), de la enfermedad mixta del tejido conjuntivo (EMTC), de la dermatomiositis y/o polimiositis (DM-PM) en casos excepcionales del lupus eritematoso generalizado (LEG), en el pseudoxantoma elástico, pero no se ha logrado demostrar de manera definitiva una alteración a nivel del metabolismo del calcio, del fósforo, de la vitamina D, de la paratohormona, ni la calcitonina, que explique una calcinosis universal5,6.
La calcinosis puede ser circunscrita, generalizada o pseudotumoral, idiopática o estar asociada con algunas de las enfermedades del tejido conjuntivo. De acuerdo con el tejido, la calcinosis puede localizarse en la piel (superficial), en el tejido celular subcutáneo de las áreas de extensión de las articulaciones (codos, rodillas), o depositarse en tejidos más profundos (músculos), como ocurre en la calcinosis universal. La calcinosis puede localizarse en tejidos desvitalizados como en la endocarditis infecciosa o en la tuberculosis, aunque también se ha observado calcinosis en algunas enfermedades de etiología desconocida como la microlitiasis alveolar pulmonar y la calcinosis arterial y glomerular descrita principalmente en los neonatos. Los depósitos calcáreos de cristales de apatita en las bursas o los tendones suelen ser otra modalidad de presentación de la calcificación distrófica. En efecto, en muchas ocasiones puede existir un daño previo microvascular, a veces asociado a microtraumas (como ocurre en la periartritis del hombro), pero en algunas enfermedades tales como los depósitos familiares de hidroxiapatita, no se conoce muy bien cómo se depositan estas sales cálcicas en las articulaciones5,6. Nos preguntamos por qué no ocurre una calcificación distrófica a nivel del tracto gastrointestinal a pesar de algunos procesos inflamatorios, como los que se observan en los pacientes con esclerosis sistémica progresiva o en la enfermedad mixta del tejido conjuntivo, como tampoco se observa calcinosis en otros órganos como cerebro, corazón, pulmón, riñón, sino estrictamente en piel, tejido celular subcutáneo y músculos como lo apreciamos en las colagenosis ya citadas. Existe compromiso de piel, tejido celular subcutáneo o músculos al tiempo en cada una de las colagenosis ya citadas, o cada una de estas patologías sólo compromete un área. Finalmente existe una vía común en el origen de la calcinosis localizada o universal en pacientes con ESP, EMTC, DM-PM o LEG, o se origina a través de mecanismos patogénicos diferentes5,6. En los últimos 15 años hemos recopilado todos los casos en los cuales documentamos la calcinosis tanto a nivel radiológico clínico como por histología. Al revisar nuestra casuística encontramos 51 pacientes con calcinosis localizada y 3 pacientes con calcinosis universal.
MATERIAL Y MÉTODOS
Se revisaron las historias clínicas y los pacientes que asistieron a la consulta externa del Hospital San Juan de Dios de Bogotá desde 1967 hasta 1996 y a la Clínica de Fracturas de Barranquilla desde enero de 1985 hasta diciembre de 1999. Se estudiaron aproximadamente 475 pacientes (250 con lupus eritematoso sistémico, 160 con esclerosis sistémica progresiva y CREST, 50 con dermato/polimiositis y 15 con enfermedad mixta del tejido conjuntivo). Todos llenaron los criterios de la ACR (American College of Rheumatology) para las enfermedades mencionadas. Se documentaron 51 pacientes con calcinosis distróficas, 48 con calcinosis localizada y 3 con calcinosis universal. De los 48 pacientes con calcinosis localizada, 40 tenían esclerodermia (25 con la forma difusa y 15 con CREST) y en ellos la calcinosis estaba localizada en áreas periarticulares de las manos; tres pacientes tenían DM y 2 PM, con calcinosis localizada a nivel paraarticular y a nivel muscular. Tres pacientes con EMTC con calcinosis localizadas a nivel articular especialmente en las manos. Todos estos hallazgos ya han sido descritos en la literatura. En vista de que esto no aporta nada nuevo al conocimiento de la calcinosis, sólo informamos de tres pacientes con calcinosis universal: una paciente con ESP con 15 años de seguimiento, un LEG con 10 años de seguimiento y una paciente con DM con 5 años de seguimiento.
Por las características clínicas, por la evolución de su enfermedad y sus diferencias histopatológicas, planteamos que las calcinosis universalis pudiesen representar tres procesos diferentes a nivel de la inflamación, pero una vía final común a nivel del depósito calcáreo (calcinosis distrófica).
PRESENTACION DE LOS CASOS
CASO 1
Presentamos el caso de una mujer de 16 años natural y procedente de Montería. La enfermedad se inició a los 7 años (en 1989) de edad con pérdida de la fuerza muscular, cansancio, dificultad para levantarse y para subir escaleras. Simultáneamente notaba la presencia de lesiones eritematosas levantadas a nivel de interfalángicas proximales (IFP), metacarpofalángicas (MCF) (pápulas de Gotron), y además lesiones de tipo violáceo alrededor de los ojos. Durante 2 años la paciente presentó la sintomatología pero no tuvo ningún diagnóstico. Ocho meses después la paciente presentó dolor y aumento de volumen de los tobillos y al examen físico se encontró un soplo sistólico GII/VI en foco mitral y se pensó en una fiebre reumática. Se practicó un ecocardiograma y se le documentó un prolapso de la válvula mitral.
Sólo hasta el 26/08/1991 el médico tratante le encuentra una disminución de la fuerza en miembros superiores e inferiores (G II/V, Gowers positivo y las pápulas de Gotron y el signo de heliotropo en párpados). Se le hace un diagnóstico de impresión: dermatomiositis, que se confirmó por histopatología y electromiografía. Se le inició tratamiento con prednisona 1 mg/kg/día, casi 3 años después del inicio de los síntomas, en ese momento la niña tenía un peso de 30 kg y una talla de 132 cm, y recibió 40 mg de prednisona en dosis dividida. En una evolución de su enfermedad narrada en su historia clínica se escribe «la paciente ha recuperado por completo la fuerza en cada segmento, pero tiene los efectos de los esteroides como el síndrome de Cushing y la presencia de estrías en la cara interna del miembro inferior izquierdo y en los codos de la paciente se observan depósitos calcáreos fistulizados por lo que recibió tratamiento con dicloxacilina por una infección sobreagregada. En 1992 se le cambió la prednisona por 30 mg de deflazacort en dosis dividida. A partir de diciembre en 1992 hasta el 13 de enero de 1998 la paciente recibió tratamiento con 24 mg de deflazacort con una mejoría parcial de su fuerza muscular a nivel espinal, pero muy poco a nivel de miembros superiores y miembros inferiores que la obligó a utilizar una silla de ruedas desde julio de 1997 hasta el presente. También presenta una contractura severa de rodillas y codos. Además del deflazacort se le inició tratamiento con 100 mg de azatioprina.
Los niveles de creatingosgoquinasa (CPK) informados desde 1991 fueron normales (15,70 U, 30 U, 26 U, 91 U) y los valores de su citología hemática mostraron un síndrome anémico con valores de hemoglobina que oscilaron entre 7,8 g y 11,5 g/l.
En diciembre de 1998, la paciente consultó a la clínica de fractura de Barranquilla en silla de ruedas.
La paciente estaba consciente, orientada, con dificultad para movilizar sus miembros (superiores e inferiores) pero podía levantar el cuello. Se observa una atrofia muscular intensa en tronco, miembros superiores e inferiores, y se notaron tumefacciones de tipo calcáreo a nivel de todo el cuerpo, pero además una contractura severa especialmente de rodillas y codos.
Diagnósticos
Dermatomiositis, fibrosis muscular, calcinosis universal, amenorrea primaria y osteoporosis severa.
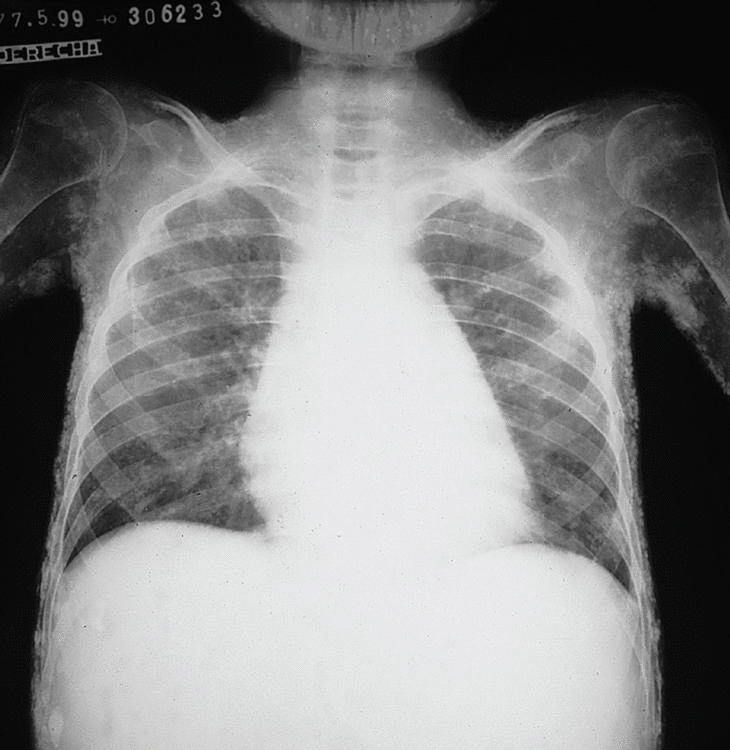
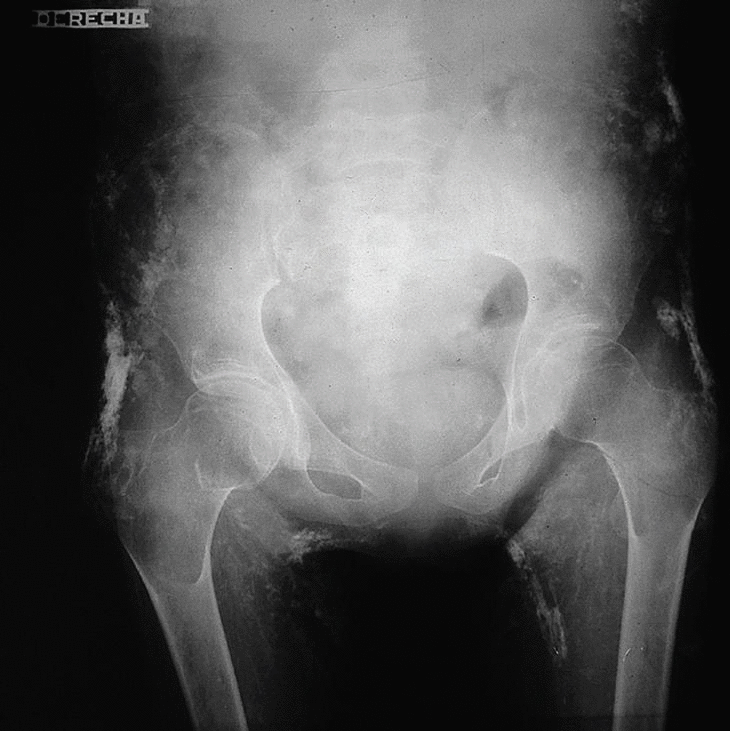
Los datos más importantes relacionados con su patología fueron: en las radiografías simples de tórax, huesos largos, pies y rodillas, se notó una pérdida de la masa muscular en las estructuras descritas y se observaron calcificaciones generalizada de todo el aparato muscular (fig. 1). En la radiografía de pelvis se observaron múltiples calcificaciones distróficas, en pared abdominal, cara interna y externa de los muslos (fig. 2).
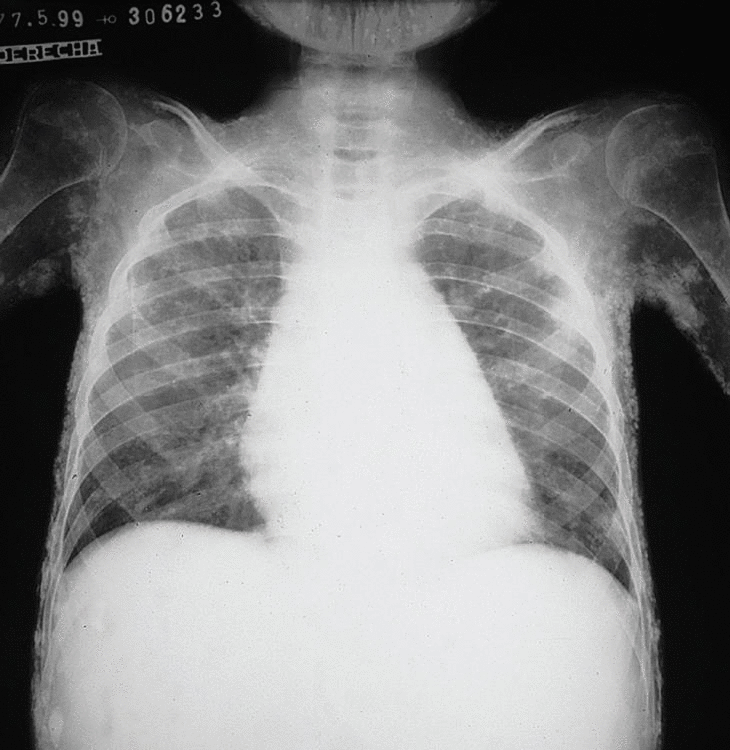
Fig. 1. Radiografía simple de tórax (A) huesos largos (B) pies y rodillas (C). Se observa una pérdida de la masa muscular en las estructuras descritas como también calcificaciones generalizadas de todo el aparato muscular.
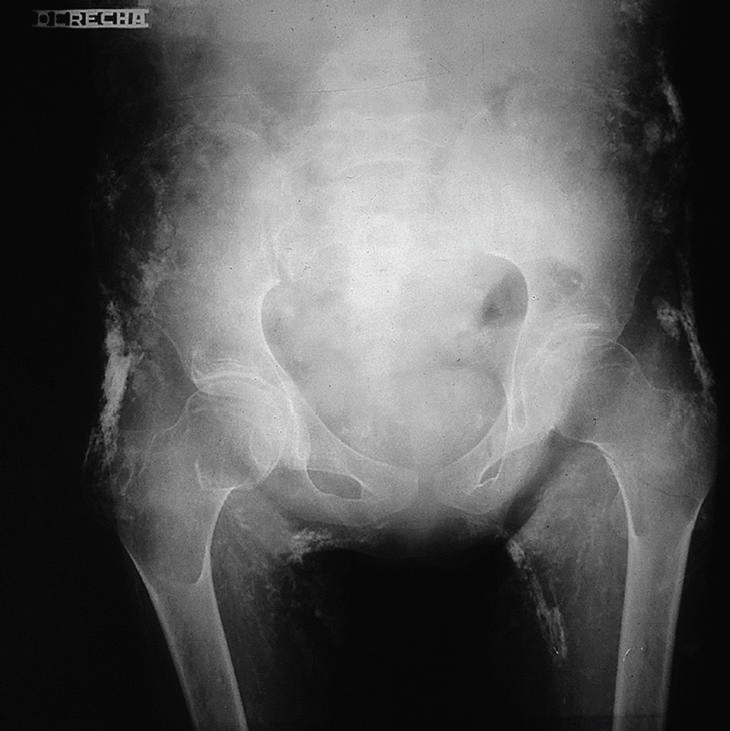
Fig. 2. Radiografía de pelvis donde se observan múltiples calcificaciones distróficas, en pared abdominal, cara interna y externa de los muslos.
La gammagrafía ósea mostró hipotrofia muscular intensa. En la densitometría ósea (22-12-1998) se evidenció osteoporosis intensa con un T-score en L2-L4 de -8,66 desviaciones estándar. La biopsia muscular y tejido celular subcutáneo documentó fibrosis intensa del tejido muscular con depósitos calcáreos.
En el laboratorio encontramos: hemoglobina de 9 g/%, hematocrito 32%, leucocitos 7.900 mm3 (neutrófilos 64%, cayados 1% eosinófilos 1%, linfocitos 30%), recuento de plaquetas 453.000/mm3, hierro sérico 26 mcg, capacidad de fijación del Fe 520 mg, porcentaje de saturación: 4,7%. El frotis de sangre periférica mostró microcitosis ++++, poiguilocitosis ++, hipocromia ++++.
Otros hallazgos fueron los siguientes: sodio: 139 meq/l, potasio: 3,7 meq/l, cloro: 102 meq/l, calcio: 4,9 mg/l, calcio: 2,0 mg/l, fósforo 3,5 mg/100 ml, calciuria: 80 mg/24 horas, fosfaturia: 800 mg/24 horas, CPK:35 U (36-188) y LDH: 458 U.
Diagnósticos finales
Calcinosis universalis secundaria a dermatomiositis; osteoporosis intensa secundaria a corticoides e inmovilización; fibrosis muscular y anemia ferropénica.
Se inició tratamiento con citrato de calcio 1,0 g, alendronato sódico 10 mg/día, calcitriol 0,25 mg/día y sulfato ferroso, pero lo más importante fue que se le suspendieron los esteroides y la azatioprina y se le inició un proceso de rehabilitación física y psicológica.
CASO 2
Se trata de una mujer, natural y procedente de Barranquilla, de 24 años actualmente. La enfermedad se le inició a los 13 años de edad, en septiembre de 1992, con poliartritis simétrica que comprometía manos (muñecas, metacarpofalángica-interfalángicas proximales), codos, rodillas, tobillos y dedos de los pies. Además presentaba un rash generalizado, migraña hemicránea izquierda, caída del cabello y pápulas en miembros inferiores. Durante su evolución al comienzo de la enfermedad refería odinofagia.
A finales de 1993 la paciente consulta por oliguria, edema de miembros inferiores y palpebral y se le documenta una proteinuria de 1,5 g/24 horas y sedimento telescopado. Anticuerpos antinucleares 1:5.020 patrón moteado, anti-Sm positivo y anti-ADN positivo.
Una biopsia renal (No. 2109-93) mostró glomerulonefritis proliferativa difusa, grado IV según la OMS, con depósitos de IgG, IgA, IgM, C3 y C4.
Además de lo anterior la paciente consulta por alopecia y vasculitis a nivel palmar en ambas manos, astenia, adinamia y dolor torácico.
En los exámenes de laboratorio se documentó anemia, leucopenia, linfopenia y una proteinuria de 2,0 g/24, hematuria y cilindros granulosos y hemáticos. Se confirmó un diagnóstico de lupus eritematoso generalizado.
Durante los años 1993 y 1994 el problema de la paciente fue su compromiso renal, articular y el rash en piel. Se le iniciaron bolos de endoxan (ciclofosfamida) cada tres meses, un bolo diario de metilprednisolona durante tres días (500 mg en cada bolo), isoniazida (300 mg) y en 1994 sulfato de cloroquina (Aralen); llamaba la atención que al reducir la dosis de esteroides se reactivaba su cuadro articular y en un renograma que se le practicó en septiembre de 1994 se demostró un alargamiento del tiempo de tránsito, lo que nos indicó compromiso parcial de sus riñones, depuración de creatinina 76 ml/min. Debido a la poca mejoría de su cuadro articular, a pesar de utilizar 15 mg de prednisona durante un año se le añade metrotexate 7,5 mg/semanal por su problema articular y renal.
En 1995 se le documenta una peri-miocarditis lúpica, persistía algo de actividad renal (depuración de creatinina de 59,7 ml/min y una proteinuria de 478,5 mg/ 24 horas).
Hay una mejoría de su cuadro articular y cutáneo. Por intolerancia al metotrexate (diarrea), se le agregan 100 mg de azatioprina/día.
En junio de 1995 consulta por un dolor en la zona gemelar, aumento de volumen de la pierna derecha, no se documenta trombosis venosa por doppler pero la paciente nuevamente tiene lesiones vasculíticas en manos, rash en tronco y miembros inferiores, además de odinofagia y disfagia alta.
En diciembre de 1995 la paciente consulta por pequeñas úlceras en varias áreas de los brazos, antebrazos, muslos y glúteos, asociada a la poliartritis de manos, rodillas y tobillos. Mejora su nefritis lúpica, pero persiste la anemia, la leucopenia y la linfopenia. En marzo de 1996 persisten las lesiones de vasculitis en manos, tronco, glúteos y asociado a una lesión isquémica del quinto dedo de la mano derecha (necrosis de la falange distal). Además persistían la anemia y la linfopenia. Las pruebas de función hepática, colesterol triglicéridos fueron normales, calcemia de 9,7 mg y fósforo de 3,8 mg. Además de la dosis de esteroides de 30 mg, asociada a la azatioprina de 100 mg/día, cada tres meses se le aplicaba un bolo de ciclofosfamida de 500 mg.
Desde el inicio de la enfermedad la paciente presentaba títulos de anticuerpos antinucleares positivos de 1:640 patrón homogéneo y títulos de anti-ADN positivos y altos en forma persistente, y en una de sus determinaciones los niveles fueron de 3.648 UI/ml. Niveles de anticardiolipina de los isotipos IgG e IgM siempre negativos.
En diciembre de 1996 presenta una neumonitis lúpica, además está febril, con pérdida de peso y artritis de rodillas y tobillo derecho.
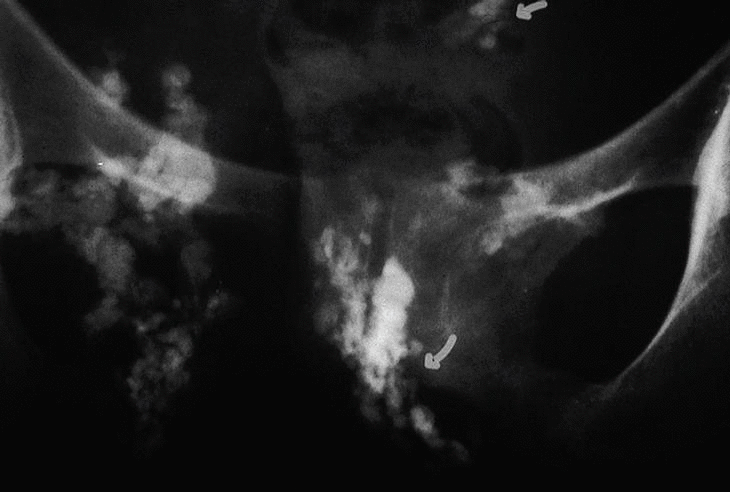
En marzo de 1997 se le observan múltiples lesiones microulcerativas en brazos, antebrazos, muslos, piernas y tronco y se plantea la posibilidad de una calcinosis generalizada. En la radiografía de antebrazos se observan claramente las calcificaciones en brazos y antebrazos. La radiografía de pelvis muestra calcificaciones (fig. 3).
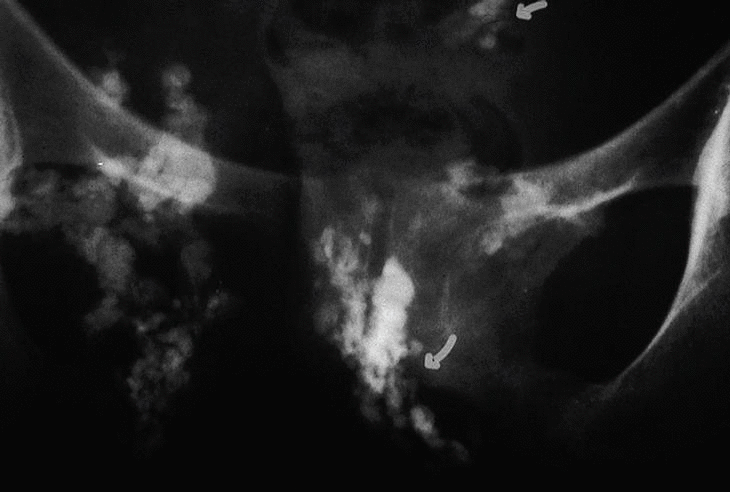
Fig. 3. Radiografía de pelvis que muestra calcificaciones distróficas.
En una ultrasonografía de calcáneo se le documentó una osteopenia secundaria a esteroides. En una biopsia de piel se documenta la calcinosis, granuloma a cuerpo extraño asociada a inflamación aguda y crónica severa.
En julio de 98 la paciente presenta una recaída de su nefropatía dada por proteinuria de 7,1/24 horas, pero una mejoría de sus lesiones cutáneas.
En marzo de 1999 persiste la nefropatía pero llaman la atención las lesiones ulceronecróticas que comprometen el cuero cabelludo, por lo que se decide incrementar la dosis de esteroides a 30 mg de prednisona diaria.
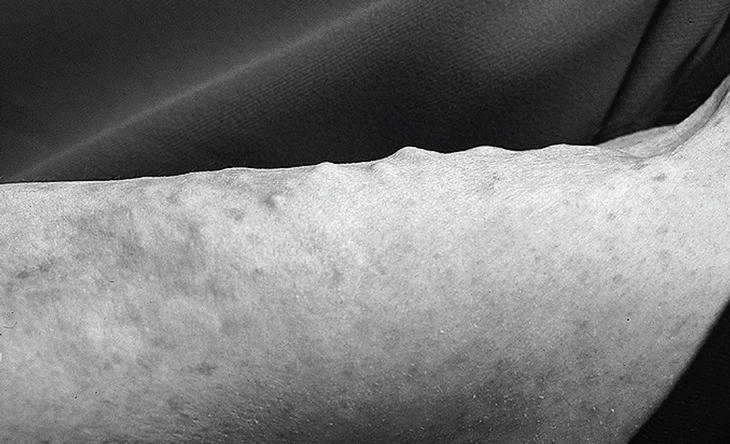
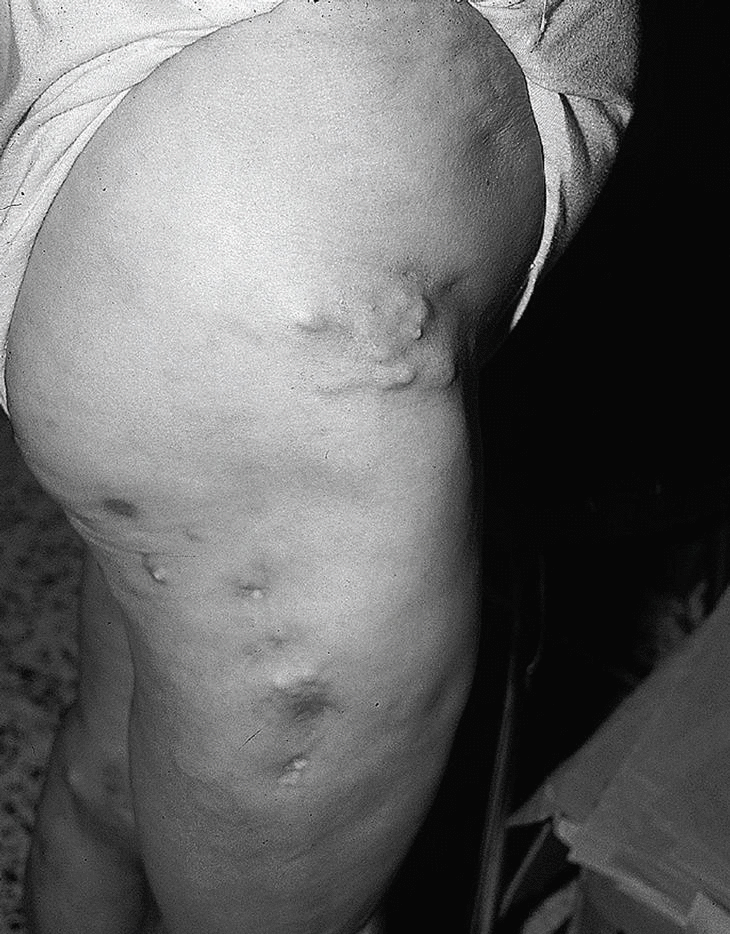
Actualmente la paciente se encuentra en remisión, pero llaman la atención las lesiones nodulares en todo el tronco, glúteos y miembros compatible con depósitos calcáreos de calcinosis universalis (fig. 4 ).
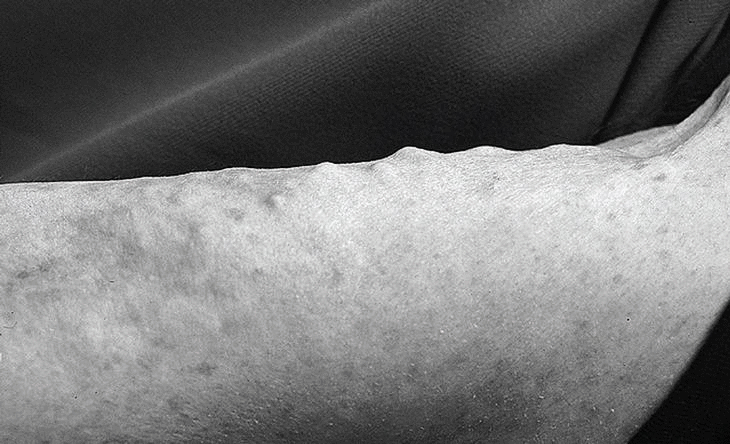
Fig. 4. Lesiones nodulares en miembros superiores.
CASO 3
Se trata de una paciente de 64 años, sexo femenino, nacida y residente de Barranquilla, que consultó el día 14 de abril de 1986 al doctor porque desde 1984 notó algunas tumefacciones en la piel, adelgazamiento y angustia. Las primeras tumefacciones aparecieron en el surco interglúteo y en los glúteos mayores; posteriormente aparecieron a nivel de la piel del trocánter mayor en forma simétrica y en la cara lateral de los muslos.
Seis meses después aparecieron las tumefacciones en las rodillas (cara anterior y posterior), codos (a nivel del olécranon), pies y piernas (cara anterior y posterior). No se observaron en la cara interna de los muslos, ni en el tronco, excepto una pequeña tumefacción a nivel del omoplato derecho. En los dos últimos años se han fistulizado a la piel con expulsión de un material blanquecino especialmente en los glúteos, codo izquierdo y cara anterior de la pierna izquierda.
Antecedentes
En 1962 a la paciente se le diagnosticó una enfermedad de Graves-Basedow y se le inició tratamiento con tapazol 45 mg/día, en dosis dividida y lugol (yoduro de potasio). Durante dos años le practicaron ese tratamiento con muy poca mejoría y en 1964 le practicaron tiroidectomía subtotal. En 1986, tuvo una recaída que se caracterizó por enflaquecimiento y pánico.
Al examen físico se encontró una paciente enflaquecida y angustiada, tolerando el decúbito, PA 170/80, peso 40,7 kg; talla 1,51 metros, FC 76/min, ojos saltones, retardo palpebral, cuello normal, cardiorrespiratorio normal, abdomen sin megalias, genitourinario normal, osteoarticular normal y fuerza muscular normal.
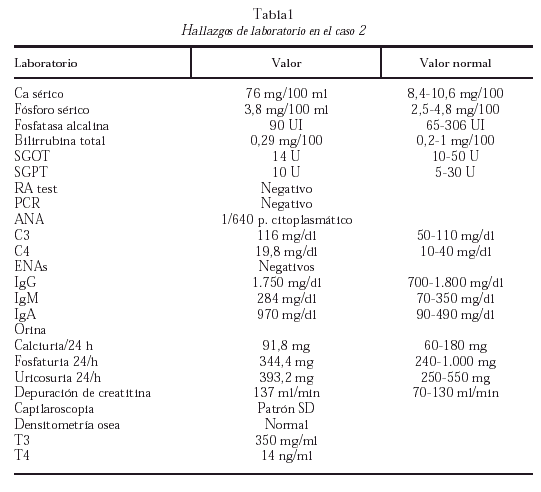
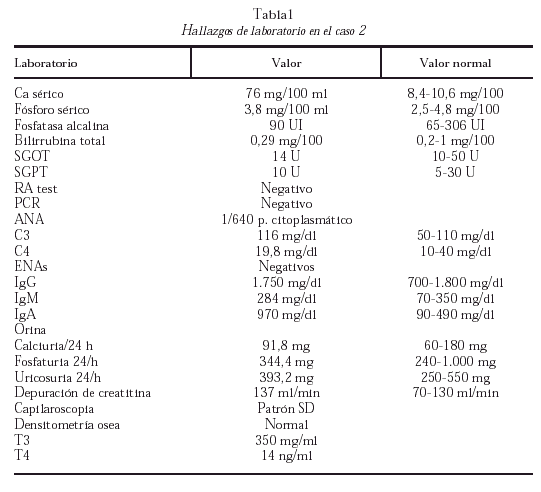
La química sanguínea encontrada se observa en la tabla 1.
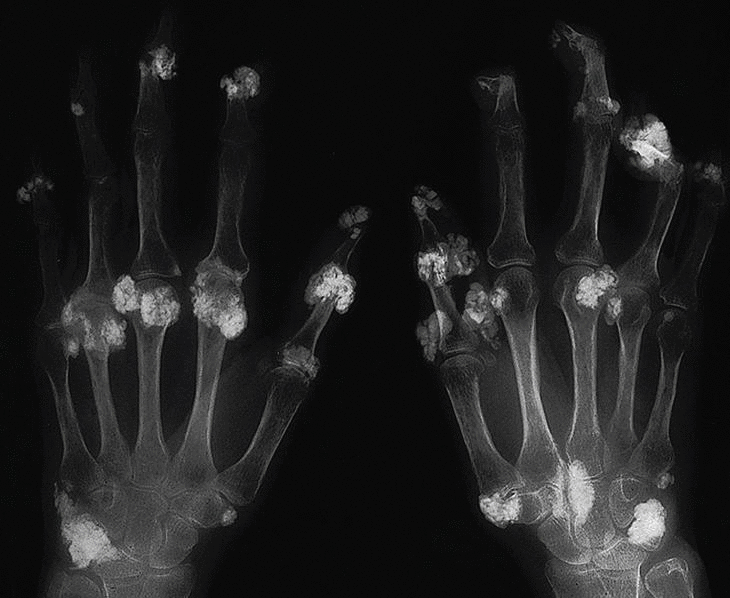
En la piel se aprecian tumefacciones y cúmulos de calcio en forma de nódulos subcutáneos en codos, rodillas, regiones trocantéricas, cara lateral de muslos, cara anterior de piernas, pies, omoplato derecho, sacro y glúteos (fig. 5). Se observó fistulación en codo izquierdo y cara anterior de la pierna izquierda, que drenaba un material blanco-amarillento. En la radiografía de manos de la paciente se observan cúmulos de depósitos calcáreos a nivel de manos (interfalángicas distales [IFD], metacarpofalángicas [MCF] y carpos) (fig. 6).
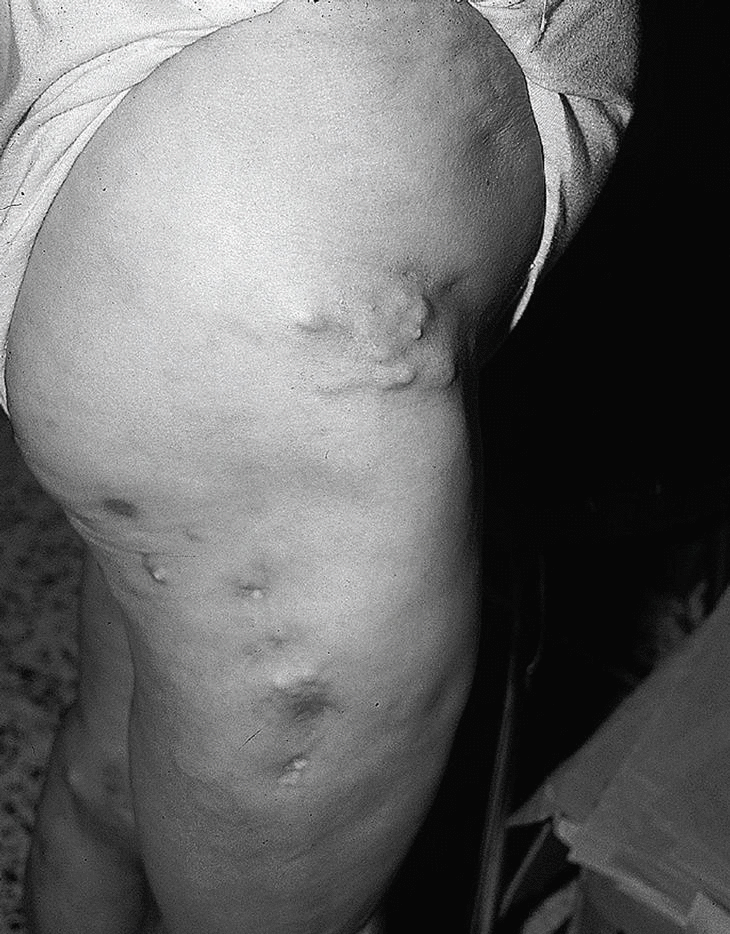
Fig. 5. Tumefacciones y cúmulos de calcio en forma de nódulos subcutáneos en regiones trocantéricas, cara lateral de muslos y cara anterior de piernas.
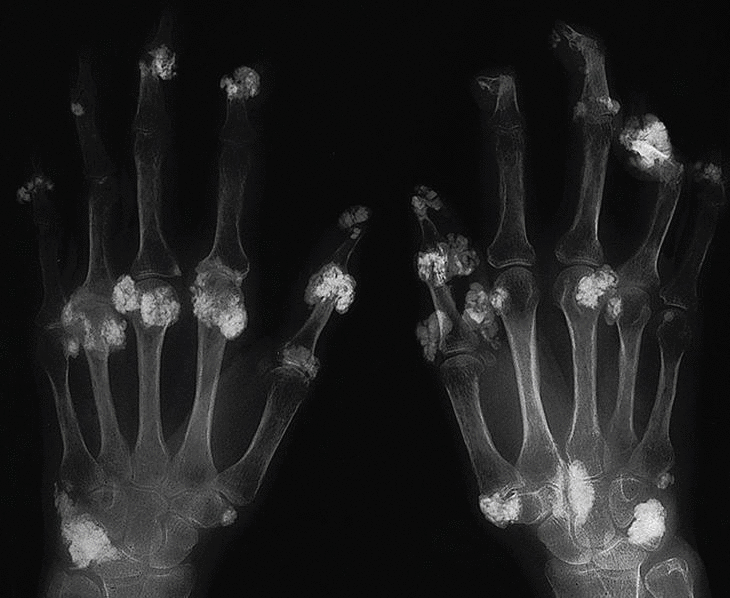
Fig. 6. Radiografía de manos de la paciente donde se observan cúmulos de depósitos calcáreos a nivel de las manos (interfalángicas distales, metacarpofalángicas y carpos).
La impresión diagnóstica fue: hipertiroidismo recidivante, hipoparatiroidismo postquirúrgico y síndrome de Thibierge- Wessenbach (esclerodermia más calcinosis).
En los estudios paraclínicos encontramos que la citología hemática, la química sanguínea, pruebas inmunológicas y las pruebas de funcionamiento renal fueron normales. Una biopsia de piel mostró: epidermis de grosor normal, cúmulo de colágeno y fibrosis de hipodermis y de la dermis que atrofia anexos y vasos y cúmulo masivo de calcio en toda la dermis. Con estos hallazgos se hizo un diagnóstico histológico de escleroderma y calcinosis cutis.
Con base en el diagnóstico de escleroderma sin escleroderma se le inició tratamiento con colchicina 1,0 mg/día y diltiazem, 120 mg/día.
DISCUSIÓN
La calcinosis cutis es un término que se utiliza indiscriminadamente para describir a un grupo de enfermedades en el cual la vía final es el depósito de sales calcáreas en la piel, este depósito a nivel de la piel fue observado inicialmente por Virchow en 1855. Las clasificaciones de la calcinosis cutis son iguales en la mayoría de los textos de enfermedades metabólicas y en los artículos especializados en estos tópicos y siempre se clasifican en cuatro tipos: metastásico, distrófico, iatrogénico e idiopático. Muchas veces los términos distróficos o idiopáticos se confunden y lo único que realmente se ha logrado identificar adecuadamente son las calcinosis distróficas, secundarias a varias patologías (tabla 1). Las calcinosis distróficas o idiopáticas a su vez la dividen en calcinosis cutis circunscrita, calcinosis cutis universalis y la calcinosis tumoral5-14.
Los mecanismos fisiopatológicos para explicar los depósitos insolubles de calcio son secundarios a factores locales (tejidos necrosados e inflamados) y a factores sistémicos. Casi siempre los depósitos calcáreos son de fosfato de calcio amorfo o cristales de hidroxiapatita. Los factores sistémicos implicados en la calcinosis cutis generalmente son de tipo metabólico y casi siempre ocurre por existir una hipercalcemia o una hiperfosfatemia, cuando el producto calcio-fósforo excede a una concentración de 70 mg2/dl2, sin que exista un tejido lesionado, sino por efecto del incremento de los niveles de paratohormona, o al péptido relacionado con la paratohormona, ocasionando un incremento extracelular de calcio-fósforo, que deriva hacia un incremento a nivel intracelular, nucleación de los cristales y precipitación cristalina, ocasionando la calcificación ectópica de los pacientes con hiperparatiroidismo, o por los tumores productores del péptido relacionado con la paratohormona5,6,14. No encontramos informes de calcinosis universalis asociado a estas patologías. Pero al revisar la literatura, es difícil encontrar informes sobre calcinosis universalis y las descripciones casi siempre están relacionadas con la dermato/polimiositis15-25 y a la esclerodermia26-30, muy pocos informes existen sobre lupus31-41 y sobre enfermedad mixta del tejido conjuntivo42-44. Nuestros casos de colagenosis con calcinosis distróficas en manos, codos, rodillas, pies y brazos son similares a las descripciones hechas en los diferentes informes de la literatura, es decir, calcinosis cutis circunscrita a esas áreas articulares asociada al daño tisular regional, que al producirse la desnaturalización de las proteínas se unen preferencialmente al fosfato y luego las sales de calcio se unen al fosfato señalando la precipitación del fosfato de calcio, en estos tejidos desvitalizados a nivel del tejido celular subcutáneo de los pacientes con esclerodermia y dermatomiositis, pero en los pacientes lúpicos existen pocos informes y los que existen no explican los mecanismos para analizar la calcinosis distrófica localizada, y menos aún explicar la calcinosis universalis45. Los tres casos que informamos en este artículo tienen una característica común y es el comienzo gradual de la calcinosis cutis, casi siempre asintomáticos excepto en la paciente con lupus. La paciente con dermatomiositis y la paciente con esclerodermia se encontraban asintomáticas o en remisión, a diferencia de la paciente con lupus que tenía un lupus muy activo. No existe una predisposición genética en estos pacientes con calcinosis universalis y sólo a nivel racial se ha observado que la calcinosis tumoral es más común en la raza negra de herencia surafricana, y de todas las calcinosis cutis es la que más se informa en la literatura universal14. En la calcinosis cutis universalis idiopática, de acuerdo a los pocos informes que existen en la literatura, no es claro el concepto de idiopático y son tan escasos los informes que no se puede descartar que sean secundarios a una colagenosis46-56.
La primera descripción del término calcinosis se utilizó en 1876 para Teissier3 en una mujer de 21 años, que desde niña tenía depósitos calcáreos en diferentes sitios de su cuerpo. Weber4 dos años después, la asocia a una esclerodermia y es la primera descripción de la asociación de calcinosis y esclerodermia, probablemente el caso de Teissier sea una polimiositis. La subdivisión de circunscrita y universal se empezó a plantear por Reines57 en 1907, fue una subdivisión arbitraria pero describía la distribución de los depósitos calcáreos en calcinosis localizada y universalis. El reconocimiento en la literatura universal de la asociación entre esclerodermia y calcinosis la hizo Thibierge y Weissenbach58,59 en 1911. Posteriormente se describen algunos casos en niños con calcificaciones en el tejido celular subcutáneo por Weber52 en 1913, Morse60 en 1921, Tisdall y Erb61 en 1924, Wilens y Deby17 en 1926, Baner et al20 en 1931 quienes describieron en la mayoría de los casos depósitos calcáreos en tejido celular subcutáneo y en los músculos. La segunda descripción de calcinosis localizada en una esclerodermia fue realizada por Thibiérge y Weissenbach58-59 en 1911, 12 años después Langme52 en un artículo clásico planteó la relación de calcinosis en pacientes con esclerodermia, dermatomiositis y en una entidad que él denomina miositis fibrosa. Posteriormente en 1931 Steinitz63 hizo una revisión exhaustiva sobre calcinosis, incluyendo sus casos y analizó algunas publicaciones por Rohstein y Welt48 en 1936, Atkinson y Weber64 en 1938, Label y Madsen16 en 1947, Wheiler6 en 1957, Kilburn47 y Sunderman49 en 1957, pero que en la mayoría de estas descripciones fueron calcinosis intersticiales, algunas asociadas a enfermedades del tejido conectivo y otras «idiopáticas».
A pesar de las pocas publicaciones sobre estos hallazgos, Tuffanelli observó y analizó el tiempo en aparecer la calcinosis en la esclerodermia y documentó que en sus casos la calcinosis aparece 11 años después del inicio de la enfermedad y la encontró en el 9% de sus pacientes65.
En la mayoría de los casos de esclerodermia, el compromiso cutáneo (acartonamiento de la piel) suele ser la primera manifestación de la enfermedad, y el compromiso visceral aparece años después. La esclerosis sistémica sin esclerodermia fue originalmente descrita por Rodnan66 en 1961, quien informa de cuatro pacientes que murieron por compromiso multiorgánico secundario a esclerosis sistémica, pero con compromiso de piel mínimo o ausente; dos de los pacientes tenían compromiso cardíaco y dos gastrointestinal. Lomeo et al67, informaron de seis casos que iniciaron con compromiso pulmonar y luego desarrollaron esclerodermia en un lapso de cuatro meses a siete años. Se adicionan cuatro casos más a la literatura con las mismas características, un importante porcentaje de estos pacientes presentaban dismotilidad esofágica, enfermedad pulmonar obstructiva, fenómeno de Raynaud, anticuerpos antinucleares positivos y capilaroscopia anormal. Posteriormente, otros informes han aparecido en la literatura.
Nuestra paciente con esclerodermia sin esclerodermia presentó como manifestación inicial de la enfermedad: calcinosis distróficas universalis sin fenómeno de Raynaud, telangiectasias, compromiso de piel o dismotilidad esofágica que sugirieran un síndrome de CREST; en su evolución no ha presentado artralgias ni compromiso de manos (8 años después).
La calcinosis, especialmente la cutis o circunscrita, se ha considerado como un hallazgo clínico importante de la esclerodermia. Su relación con la variante CREST ha sido bien estudiada y en especial su fuerte asociación con los anticuerpos anticentrómero (ACA) en pacientes con enfermedades del tejido conectivo ha sugerido que éstos son un factor de riesgo para el desarrollo de calcinosis. La incidencia de calcinosis en pacientes con ACA varía del 42% al 71%. En el estudio de Itoh y Sato68 se encontró asociación entre HLA-A2 con la calcinosis, lo que sugeriría un subgrupo de pacientes genéticamente determinados con riesgo de esta complicación.
Nuestra paciente tenía anticuerpos antinucleares (ANA) positivos, patrón citoplasmático y su calcinosis era universalis. Nosotros reportamos recientemente seis pacientes con calcificaciones distróficas generalizadas que tenían dermatomiositis del adulto pero sin calcinosis cutis. El caso que reportamos ahora es compatible con esclerodermia en la biopsia de piel y no tiene ningún hallazgo de miopatía inflamatoria. La calcinosis universalis ha sido descrita en niños con dermatomiositis, usualmente cursa con vasculitis sistémica, los estudios histopatológicos revelan alteraciones en la microvasculatura similares a las encontradas en la esclerodermia, sugiriendo un mecanismo fisiopatológico común que compromete grandes áreas del cuerpo, que ocasiona isquemia tisular, ya sea por un proceso inflamatorio vascular o por una vasculopatía. Usualmente los niveles de calcio y fósforo son normales. Nuestra paciente tenía discreta hipocalcemia y antecedentes de cirugía de tiroides, pero todos los estudios relacionados con metabolismo óseo son normales, lo que sugiere un mecanismo de calcinosis distrófica.
Otros diagnósticos diferenciales a considerar en la calcinosis universalis incluyen la calcinosis tumoral, la insuficiencia renal crónica, hipervitaminosis D3, síndrome leche-alcalino, enfermedad por microcristales, hiperparatiroidismo, etc. Algunos clasifican las calcificaciones cutáneas en cuatro tipos: distróficas (como en nuestro caso), idiopática, tumoral y metastásica. Se han descrito en lupus eritematoso sistémico (LES), pacientes con insuficiencia renal crónica bajo tratamiento con hemodiálisis probablemente por hiperfosfatemia e hiperparatiroidismo secundario, mecanismo diferente al de los pacientes parapléjicos que desarrollan calcificaciones distróficas por irritación local. También se han descrito estas calcificaciones en las conjuntivas de los pacientes renales y se ha observado que probablemente la intoxicación por aluminio sea igualmente un factor calcificante5,6.
Las de origen tumoral ocurren por calcificaciones con formación de hueso, por metaplasia de tejido cartilaginoso y por necrosis, hemorragias o cambios de los tejidos. Otras calcificaciones son del tipo distrófico, como las observadas en los seminomas. Otras causas de calcinosis incluyen enfermedades granulomatosas, procesos infecciosos y aplicación local de glucocorticoides5,6.
La paciente que informamos pertenece al subgrupo de esclerosis sistémica sin escleroderma y para algunos hace parte de la forma limitada. Según Leroy et al69, su diagnóstico se basa en fenómeno de Raynaud, dismotilidad esofágica, telangiectasias, hallazgos capilaroscópicos y compromiso pulmonar. Sus características clínicas, serológicas y el pronóstico son menos conocidos. La capilaroscopia es muy útil en su sospecha. La biopsia de piel en fases tempranas revela atrofia epidérmica, fibrosis dérmica, aumento de mastocitos e infiltrado monuclear perivascular. La presencia de calcinosis universalis en esclerodermia no ha sido descrita previamente a diferencia de la forma circunscrita que se encuentra más frecuentemente en el síndrome de CREST. Lally et al69,70 analizaron 91 pacientes con esclerodermia y encontraron que el 14% se presentaban con manifestaciones atípicas, dentro de las cuales no se incluía la calcinosis; al igual que la ausencia de su mención en revisiones recientes. La asociación de esclerosis sistémica sin esclerodermia con LES ha sido previamente informada, caracterizándose por presencia de fenómeno de Raynaud, dismotilidad esofágica y fibrosis pulmonar.
La calcinosis universalis es una entidad que desde las descripciones clásicas y de acuerdo a los sitios donde se acumula el depósito calcáreo la clasifican en circunscrita, «idiopática» o asociada a las enfermedades del tejido conjuntivo. La forma idiopática descrita en un niño en 1931 por Bauer et al55, se relacionó con un cuadro de dermatomiositis, lo mismo observó con el artículo descrito por Leystina et al56, en 1964, sobre calcinosis intersticial, ya que en estos artículos no se plantea en forma clara su mecanismo patogénico, sino que se realiza una descripción de los depósitos de las sales de calcio en el tejido celular subcutáneo. En la primera edición del libro de Resnick y Niwayama71, en 1981, la calcinosis universalis idiopática es un resumen de los artículos mencionados anteriormente. Posterior a esta descripción de la forma idiopática, no encontraron documentación en la literatura médica. En nuestro criterio para que se induzca una calcinosis es necesario que exista un tejido lesionado y esto predispone a la calcificación extraesquelética asociada a un producto de solubilidad sérica calcio-fósforo normal.
La lesión tisular que se lleva a cabo en la dermis y en el tejido celular subcutáneo, en los músculos, ligamentos, tendones, bursa, suele ser secundario a un proceso inflamatorio, necrótico, fibrótico, infeccioso, hemorrágico, que puede liberar un material que tiene las propiedades de nuclear sales de fosfato de calcio y carbonato de calcio que se depositan en estos tejidos produciendo nódulos calcáreos que van a coalecer ocasionando grandes masas y por ende la calcificación distrófica. De acuerdo al tamaño del depósito de sales de calcio y del sitio del depósito, la calcinosis distrófica que se observa en la dermato/polimiositis, síndrome de CREST, esclerosis sistémica progresiva son la calcinosis circunscrita a la dermis y tejido subcutáneo en las falanges distales, o una extensa en los dedos (calcinosis digital), alrededor de la articulación como se observa en la enfermedad mixta del tejido conectivo, dedos, palma de las manos, en antebrazos, muslos, piernas y alrededor del trocánter. La calcinosis tumoral que es el segundo tipo de calcificación de acuerdo a la clasificación de Greenfield72, se ha descrito asociada a dermato/polimiositis. Generalmente son asintomáticas, pero cuando los depósitos se presentan en áreas de roce o trauma como en los codos, o en áreas que soportan peso como en los glúteos son dolorosas. En una de nuestras pacientes con dermatomiositis encontramos masas secundarias a depósitos calcáreos en glúteos y muslos que condicionan una calcinosis distrófica pseudotumoral.
De acuerdo a la clasificación de Greenfield72, publicada en 1975, se clasifica el depósito de las sales de calcio en los tejidos blandos en tres tipos: calcificación metastásica relacionada a una alteración en el metabolismo del calcio y del fósforo, calcinosis secundaria al depósito de sales de calcio en el tejido subcutáneo en presencia de un metabolismo calcio-fósforo normal y calcinosis distrófica cuando las sales de calcio se depositan en el tejido desvitalizado o dañado en ausencia de un trastorno metabólico.
En cuanto al lupus en un informe de Rothe, Grant-Kels y Naomi Rothfield41, informan un caso de calcinosis extensa cutis y revisan otros 23 pacientes con estas mismas características que coinciden en que los pacientes con lupus que se complicaron con una calcinosis extensa cutis tenían una enfermedad con una larga evolución. Marzano et al73 describieron un caso de calcinosis distrófica en un paciente con lupus cutáneo subagudo.
En conclusión, podemos plantear que las calcinosis univesalis asociadas a las diferentes colagenosis tienen mecanismos patogénicos diferentes, pero asociadas generalmente a un proceso inflamatorio que deja un sustrato tisular lesionado ocasionando la calcinosis distrófica que es similar en todas las colagenosis estudiadas en éste artículo.
NOTICIAS
OSTEOPOROSIS SECUNDARIAS
VII REUNIÓN MONOGRÁFICA DE LA SOCIEDAD ESPAÑOLA DE INVESTIGACIÓN ÓSEA Y METABOLISMO MINERAL (SEIOMM)
Y
IV CURSO NACIONAL DE LA FUNDACIÓN HISPANA DE OSTEOPOROSIS Y ENFERMEDADES METABÓLICAS ÓSEAS (FHOEMO)
Con el patrocinio de la International Osteoporosis Foundation (IOF)
Toledo, 3 y 4 de octubre 2002
Información: Pharmacongress
Avda. de Burgos, 12 28036 Madrid