




La enfermedad de Pompe (EP) de inicio tardío es una alteración autosómica recesiva que presenta una miopatía a predominio de miembros inferiores y compromiso de los músculos respiratorios. Reportamos un caso de EP de inicio tardío con retraso diagnóstico debido a la presencia de dolor tipo fibromialgia.
Caso clínicoLa paciente presentaba una larga historia de dolor tipo fibromialgia que no mejoró con pregabalina. En el examen físico, se observaron 15 puntos de sensibilidad junto con debilidad en miembros inferiores. El examen funcional respiratorio evidenció una caída de la capacidad vital forzada del 19,5% en el decúbito y normal en posición supina, indicativo de compromiso diafragmático. En el laboratorio, los niveles de creatina-fosfocinasa se encontraban levemente elevados, siendo el test de gota seca positivo para la deficiencia de la maltasa ácida. Se confirmó el diagnóstico mediante evaluación de los niveles a nivel leucocitario.
ConclusionesEs necesario tener presente esta presentación atípica de la EP de inicio tardío, ya que puede aparejar retrasos en el diagnóstico y tratamiento.
Late-onset Pompe¿s disease is a rare autosomal recessive disorder which usually presents as a limb-girdle myopathy with early respiratory involvement. We report a case of a delayed Pompe's disease diagnosis in a patient with fibromyalgia-like pain.
Clinical caseThe patient had a long history of fibromyalgia-like pain without improvement with pregabalin. At clinical examination, 15 sensitive points were observed along with lower extremity weakness. Pulmonary function test revealed normal values upright and a 19% reduction in vital capacity with recumbent position, suggestive of diaphragmatic compromise. The serum creatine kinase level was mildly elevated and a dried blood spot test indicated acid maltase deficiency. The diagnosis of late-onset Pompe's disease was confirmed by leukocyte test of acid maltase deficiency.
ConclusionsClinicians should be aware of this atypical presentation of late-onset Pompe's disease to avoid unnecessary delays in diagnosis and treatment.
La glucogenosis tipo ii o enfermedad de Pompe (EP) es una enfermedad poco frecuente, autosómica recesiva, generada por la ausencia o deficiencia severa de la proteína lisosomal alfa glucosidasa ácida cuya función es desramificar los enlaces 1,4 y 1,6 de la maltosa y residuos de glucógeno que ingresan a los lisosomas para formar glucosa. Este déficit enzimático da lugar a la acumulación de glucógeno lisosomal, principalmente del tejido muscular y cardiaco, provocando el daño de las membranas lisosomales y la liberación masiva de glucógeno hacia citosol de estos tejidos, entorpeciendo su función contráctil y resultando en hipotonía y debilidad muscular progresiva.
La enfermedad posee una amplia gama de fenotipos clínicos con grados variables de progresión, síntomas de inicio y niveles de afectación de los distintos órganos1, reconociéndose 2 extremos: una forma de inicio infantil de rápida progresión y otra de inicio tardío con progresión más pausada2,3.
Su clínica proteiforme constituye un verdadero desafío diagnóstico4-6, siendo primordial su identificación temprana, ya que se cuenta con terapia de reemplazo enzimático que ha demostrado modificar la historia natural de la enfermedad7. Reportamos un caso de EP con sintomatología atípica que presentó demora diagnóstica debido a ser diagnosticada inicialmente como fibromialgia.
Caso clínicoMujer de 52 años de edad, remitida desde Atención Primaria por dolores musculares generalizados a predominio de ambos muslos y región lumbar, de aproximadamente 15 años de evolución, que empeoran con la actividad física e interfiere con las actividades de la vida diaria. Relata una dificultad progresiva para subir las escaleras y fatiga con caídas frecuentes, siendo valorada por un médico ortopedista que le receta plantillas. Ocho años atrás, se le diagnostico fibromialgia, por lo cual se le indicó tratamiento con pregabalina 150mg cada 12 h. La paciente presentó una pobre respuesta al tratamiento, refiriendo en el último año insomnio de mantenimiento.
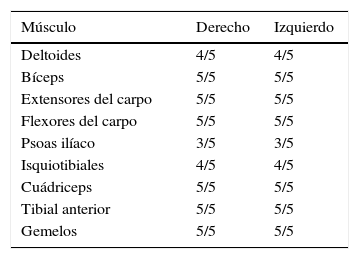
En la exploración general, se observaron 15 puntos de sensibilidad, así como una marcha lenta y basculante en postura hiperlordótica (marcha miopática). El examen de fuerza segmentaria con el uso de la escala de la Medical Research Counsil (0 puntos: contracción muscular mínima sin movimiento a 5 puntos: fuerza normal) mostró debilidad proximal simétrica, sin compromiso de músculos cervicales, faciales o de la lengua (tabla 1). En posición de decúbito dorsal, la paciente no podía flexionar el tronco sobre los muslos, lo que indicaba una marcada debilidad en los músculos abdominales y paraespinales. No se constata escápula alada. Previo a la consulta, se le realizaron 2 electromiografías. Se evaluaron los siguientes músculos: deltoides, bíceps, supinador largo, extensor común de los dedos, extensor propio del índice, oponente del pulgar, primer lumbrical y abductor del meñique para miembros superiores y vasto externo, recto anterior, tibial anterior, gemelo interno, extensor propio del hallux y pedio en los miembros inferiores. La descripción de los músculos evaluados no reveló actividad espontánea en reposo, con patrón de reclutamiento interferencial a la contracción muscular, siendo los potenciales de unidad motora de amplitud y morfología normales.
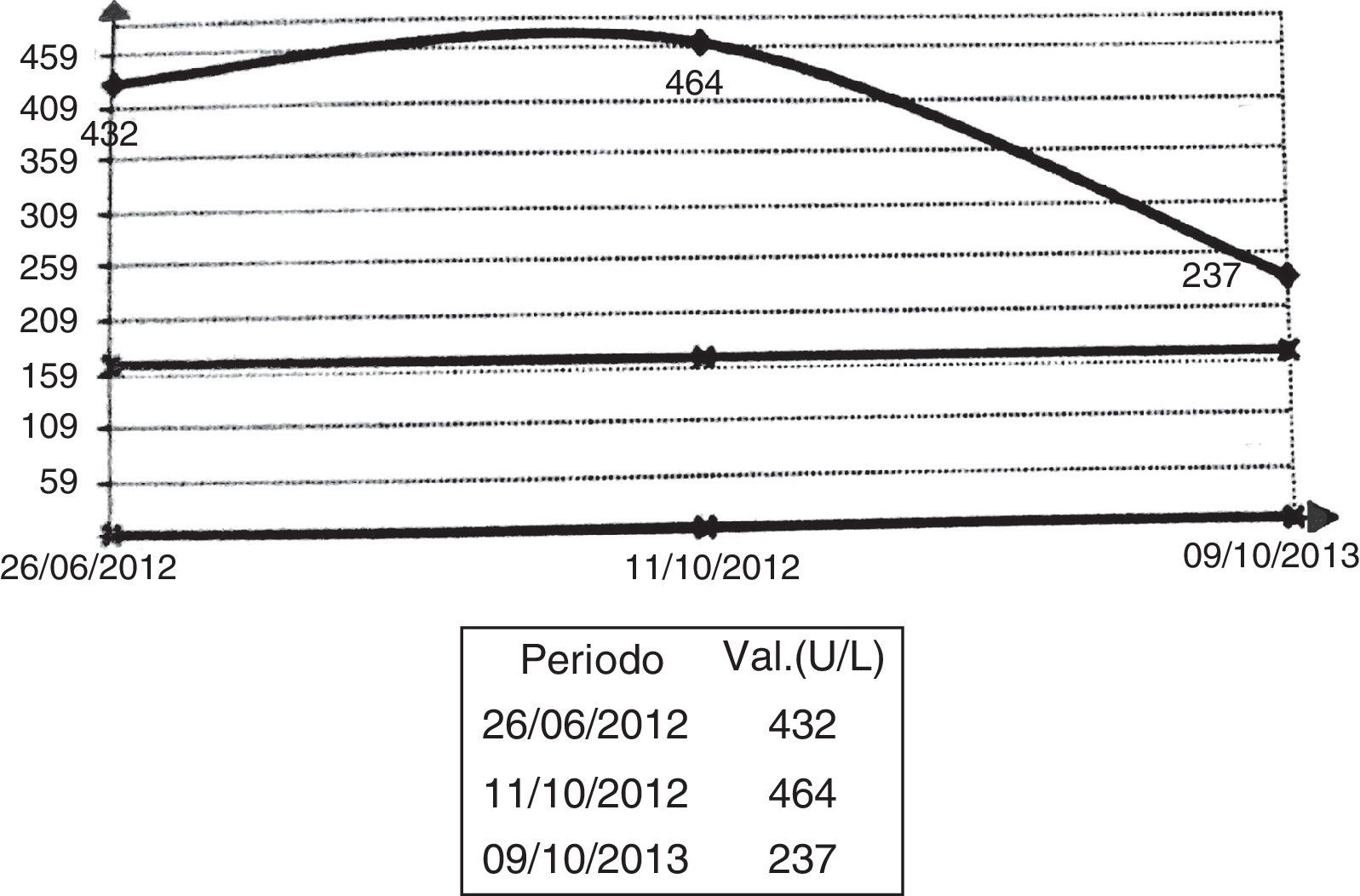
El examen funcional respiratorio evidenció una caída de la capacidad vital forzada del 19,5% en el decúbito y normal en posición supina, indicativo de compromiso diafragmático. Las pruebas de laboratorio mostraron un hemograma, un hepatograma y bioquímica normales, así como resultados negativos para autoanticuerpos (anti-Ro, anti-La, anti-DNA, anti-RNP y anti-Jo). Se observó una elevación discreta de la creatincinasa (CK): 237-437 UI (fig. 1). Debido al patrón de compromiso muscular axial y a la debilidad diafragmática indicada en el estudio espirométrico, se entretuvo el diagnóstico de EP. El dosaje de alfa glucosidasa por gota seca fue de 58,5 μmol/l/h (valor normal < 30 μmol/l/h) y el porcentaje de inhibición de la alfa glucosidasa ácida fue de 95 μmol/l/h (normal < 87, patológico > 89) compatible con la enfermedad. Se confirmó el diagnóstico mediante el dosaje de la alfa glucosidasa en leucocitos.
Comentarios
Reportamos a una paciente con EP con una larga historia de mialgias y fatiga, erróneamente diagnosticada como fibromialgia. Recientemente, se reportaron 2 casos de 2 hermanas con sintomatología compatible con fibromialgia de larga duración y quejas gastrointestinales. Similar al presente caso, la CK se encontraba elevada y los estudios electromiográficos fueron reportados como normales. A diferencia del caso, ambas presentaban fuerza normal en los 4 miembros y ausencia de compromiso diafragmático, y el diagnóstico fue sospechado luego de realizar una biopsia muscular8.
La paciente presentó un fenotipo compatible con la variante de inicio del adulto, con debilidad axial y mayor compromiso muscular en los miembros inferiores a predominio de ambos muslos y respetando el cuádriceps femoral, músculo que se ve afectado solo en estadios avanzados de la enfermedad2. Como en este caso, los pacientes con EP pueden presentar compromiso diafragmático y eventualmente insuficiencia respiratoria1,2,6. Los reflejos osteotendinosos se reducen o desaparecen solo cuando el compromiso muscular es severo2. Otras manifestaciones clínicas incluyen escoliosis, hiperlordosis, síndrome de espina rígida, trastorno del sueño, intolerancia al ejercicio, fatiga, calambres musculares y dolor lumbar. Cabe destacar que el dolor muscular y la fatiga referida inicialmente por la paciente constituyen síntomas inespecíficos que pueden observarse en una gran cantidad de enfermedades endocrinológicas, metabólicas o inflamatorias, incluyendo las miopatías. En la EP, el 76% de los pacientes presentan fatiga en la evolución de la enfermedad y un 24% lo manifiesta como primer síntoma, resultando más frecuente en aquellos con compromiso del sueño o de los músculos respiratorios9. La prevalencia de dolor muscular en esta enfermedad varía entre el 18 y el 45%, y su causa es multifactorial2,10. Una de sus causas principales es la afectación postural debido al estrés que sufre el sistema musculoesquelético, secundaria a la debilidad de muscular. Al igual que en pacientes con enfermedad de Mc Ardle o en distrofia miotónica tipo i, el agotamiento muscular también puede generar dolor muscular en pacientes con EP.
La presencia de estudios electromiográficos normales, o informados como normales, no descarta su diagnóstico, pudiéndose constatar descargas miotónicas, descargas repetitivas de alta frecuencia o potenciales de pequeña amplitud y corta duración, particularmente en los músculos paraespinales.
Niveles anormales de CK indican un compromiso muscular, aunque sus valores en la EP pueden ser normales o discretamente elevados pero sin sobrepasar 1.000 UI. En un estudio que evaluó la historia natural de la enfermedad en 225 pacientes; el 9% presentaba valores normales de CK11.
El compromiso respiratorio debido a la debilidad diafragmática se traduce en un síndrome restrictivo que condiciona el pronóstico de esta enfermedad. En el presente caso, la espirometría en posición sedente fue normal pero se evidenció una caída del 19,5% con el decúbito supino. La caída de la capacidad vital forzada ¿ 10% es indicativa de la enfermedad, especialmente en aquellos pacientes con mialgias y elevaciones de la CK. De esta forma, todo paciente con sospecha de compromiso muscular, mialgias y elevaciones de la CK debe realizarse una espirometría en sedestación y en posición decúbito supino.
La biopsia muscular no es parte del algoritmo diagnóstico de la EP, excepto en la presencia de estudios enzimáticos no concluyentes. Los estudios patológicos demuestran que un 40-80% de las biopsias musculares presentan vacuolas con tinción PAS (fosfatasa ácida positiva) compatibles con un contenido anormal de glucógeno. En la EP, las biopsias pueden ser normales, tener cambios mínimos detectables (como vacuolas PAS positiva) o bien características severas similares a las descritas en la forma infantil, con fibras necróticas y un infiltrado inflamatorio secundario, lo que ha llevado en varios casos a un diagnóstico erróneo de miopatía inflamatoria6,12. Hasta el año 2000, no se disponía de ningún tratamiento específico para esta enfermedad, por lo que se aplicaban solo medidas paliativas. En la actualidad, la terapia de reemplazo enzimático con alglucosidasa alfa humana recombinante (Myozyme®, Genzyme Corp., Cambridge, MA, EE. UU.) constituye el único tratamiento efectivo, demostrando una mejoría de la función muscular, cardiaca y respiratoria7,13,14. Particularmente en la EP de inicio tardío, dicha terapia es beneficiosa, siendo el objetivo principal la prevención de la pérdida adicional de función muscular.
Los primeros pasos para efectuar un diagnóstico precoz en la EP son: a) reconocer las posibles variantes en su presentación clínica; b) considerar esta patología dentro de los posibles diagnósticos diferenciales, y c) planificar una apropiada evaluación. Se propone el estudio sistemático de la función enzimática en gota seca en todo paciente con mialgia persistente y elevación de la CK, incluso con biopsia muscular normal. En el caso ahora reportado, la paciente fue inicialmente diagnosticada como un cuadro de fibromialgia; sin embargo, según los criterios de Wolfe15, antes deben descartarse otras patologías que expliquen el dolor muscular persistente. Ante la presencia de un niño o adulto con debilidad muscular progresiva con un patrón de compromiso axial, aun en casos donde se demuestre un biopsia muscular que pudiera arrojar un diagnóstico de miopatía inflamatoria o que sea de características normales, es necesario solicitar el dosaje enzimático de alfa glucosidasa, ya que el inicio de la terapia de reemplazo en estadios iniciales se encuentra asociada a una mejor respuesta y pronóstico a largo plazo.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
