



La ataxia de Friedreich (AF) es la ataxia degenerativa recesiva más común y la más prevalente de todas las ataxias hereditarias (2-4 casos por 100.000)1-3. Los pacientes presentan ataxia progresiva de la marcha/miembros acompañada de debilidad motora, arreflexia en miembros inferiores, pérdida de la propiocepción, disartria, nistagmo y alteraciones auditivas. Los primeros síntomas tienden a ocurrir alrededor de la pubertad, si bien su inicio puede variar desde la primera infancia (2-3 años) hasta la adultez (> 25 años), progresando hasta la pérdida de su capacidad para caminar unos 10-15 años después de la aparición de la enfermedad2,4. La afectación cardiológica ocurre en el 90-100% de los pacientes, secundaria al proceso infiltrante, y puede variar desde alteraciones menores en el electrocardiograma (ECG), hasta arritmias y alteraciones estructurales como hipertrofia del ventrículo izquierdo o fibrosis intersticial con importante proliferación colágena y degeneración grasa, que es una causa frecuente de muerte prematura en estos pacientes1,5-7.
La mayoría de los casos de AF se deben a una expansión de repetición de tripletes de la secuencia GAA en el primer intrón del gen de la frataxina (FXN) localizado en el cromosoma 9q21.111,8. La frataxina es una proteína mitocondrial que activa el ensamblaje en proteínas de grupos hierro-azufre (Fe/S) como parte de un complejo multiproteico. Aunque su función fisiológica exacta no ha sido aún establecida, se cree que tiene un papel esencial en la integridad y el funcionamiento de las mitocondrias y en el metabolismo del hierro celular2. También ha sido implicado, posiblemente de manera indirecta, en la señalización de sistemas de defensa antioxidante y en vías que controlan la supervivencia o muerte celular2,4. La deficiencia de frataxina se asocia con una alteración de la biogénesis de los grupos Fe/S, de la homeostasis del hierro y un aumento de los niveles de estrés oxidativo que en última instancia conducirá a la neurodegeneración y la miocardiopatía2,4.
Aunque existen algunas líneas de investigación sobre posibles fármacos modificadores de la enfermedad centrado en compuestos que aumenten los niveles de frataxina o disminuyan las consecuencias de su deficiencia (antioxidantes, etc.), en la actualidad no existe un tratamiento curativo para la AF9-12. Por tanto, las opciones terapéuticas se limitan a la prevención y el tratamiento de las complicaciones neurológicas derivadas de la progresión de la enfermedad (fisioterapia, terapia ocupacional, tratamiento ortopédico de la escoliosis, etc.) y de sus comorbilidades extraneurológicas, como la cardiopatía (uso de beta bloqueantes o inhibidores de la enzima conversora de angiotensina) o la diabetes.
El factor del crecimiento similar a la insulina tipo 1 (IGF-1) estimula el crecimiento y la proliferación celular e inhibe la muerte celular programada. El IGF-1 ha demostrado efectos neuroprotectores, así como efectos beneficiosos en el tratamiento de diversas enfermedades neurodegenerativas hereditarias causadas por la expansión de tripletes CAG como SCA3, SCA6 y SCA7, y en la AF en una prueba de concepto, debido posiblemente al aumento de los niveles de frataxina y a una mejoría de la actividad mitocondrial13-18. Aunque los datos son limitados, decidimos iniciar el tratamiento con IGF-1 humano recombinante (rhIGF-1) (Increlex®, mecasermina [rDNA], vial 10 mg/ml. Ipsen Pharma SA, España) en un niño con ataxia de Friedreich, dentro de un programa de uso compasivo.
Se evaluaron de forma prospectiva la seguridad y la eficacia del tratamiento con rhIGF-1 administrado por vía subcutánea (dosis de inicio: 25μg/kg 2veces al día) en una niña de 13 años con AF diagnosticada y en tratamiento con terapia antioxidante (coenzima Q10 con vitamina E, idebenona) desde los 9 años de edad, dentro de un programa de tratamiento de uso compasivo. El estudio fue aprobado por el comité ético de nuestro centro y la paciente y su familia proporcionaron su consentimiento informado.
Se elaboró un protocolo de evaluación previa al inicio del tratamiento consistente en: exploración neurológica completa, análisis de laboratorio (hematología, bioquímica, pruebas de función hepática y renal, niveles séricos de IGF-1, etc.), ECG, ecocardiografía y ecografía abdominopélvica. Tras el inicio de la terapia con rhIGF-1, se programaron visitas trimestrales durante el primer año de seguimiento y posteriormente semestrales hasta completar un total de 36 meses19. La eficacia se evaluó mediante la escala de evaluación y clasificación de ataxia (SARA)20 y se filmaron todos los ítems enumerados en la misma. Estos vídeos fueron posteriormente revisados por un revisor independiente que fue cegado de acuerdo con las fechas de los vídeos. Respecto a la seguridad del tratamiento, monitorizamos los niveles de glucosa en sangre capilar utilizando un glucómetro durante el primer mes de tratamiento y después de cada aumento de la dosis, para vigilar posibles hipoglucemias. Además, se establecieron controles analíticos, niveles séricos de IGF-1, ECG, ecocardiografía y ecografía abdominal cada 6 meses.
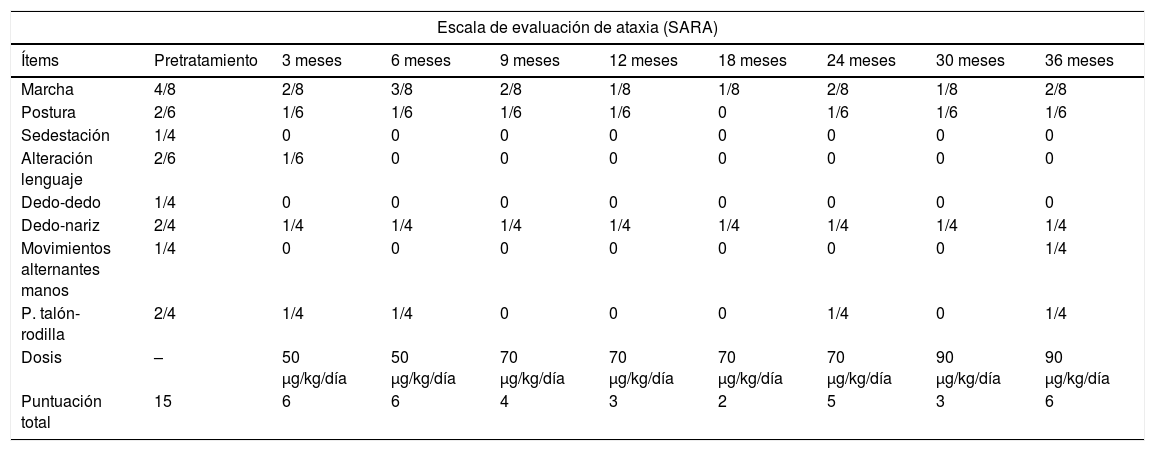
Durante un período del estudio, se observó una reducción en la progresión del deterioro neurológico. Observamos una mejoría en la marcha (patrón y velocidad), el equilibrio (disminución del número de caídas) y de la tolerancia al ejercicio. Su lenguaje se hizo más fluido y progresó en tareas relacionadas con sus habilidades motoras finas, como la caligrafía y el aumento de la velocidad de escritura. Aunque el impacto de su influencia en la calidad de vida no fue medida mediante una escala formal, subjetivamente tuvo un impacto positivo sobre la autoestima y el estado de ánimo de la paciente. Esta mejoría sintomática se evidenció desde los primeros meses del tratamiento con rhIGF-1. Se produjo un descenso progresivo en la puntuación total de la SARA desde la situación basal (15/40) hasta el final del estudio (6/40), con ciertos altibajos que precisaron el aumentar la dosis del tratamiento. La puntuación total y de cada uno de los ítems de la SARA, así como la dosis de rhIGF-1, se exponen en la tabla 1. El revisor independiente también observó una clara mejora en la coordinación motora y el equilibrio en los vídeos, y estuvo de acuerdo con nuestras calificaciones SARA.
Evolución neurológica según la escala de evaluación y clasificación de ataxia (SARA)
| Escala de evaluación de ataxia (SARA) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ítems | Pretratamiento | 3 meses | 6 meses | 9 meses | 12 meses | 18 meses | 24 meses | 30 meses | 36 meses |
| Marcha | 4/8 | 2/8 | 3/8 | 2/8 | 1/8 | 1/8 | 2/8 | 1/8 | 2/8 |
| Postura | 2/6 | 1/6 | 1/6 | 1/6 | 1/6 | 0 | 1/6 | 1/6 | 1/6 |
| Sedestación | 1/4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Alteración lenguaje | 2/6 | 1/6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dedo-dedo | 1/4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dedo-nariz | 2/4 | 1/4 | 1/4 | 1/4 | 1/4 | 1/4 | 1/4 | 1/4 | 1/4 |
| Movimientos alternantes manos | 1/4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1/4 |
| P. talón-rodilla | 2/4 | 1/4 | 1/4 | 0 | 0 | 0 | 1/4 | 0 | 1/4 |
| Dosis | – | 50 μg/kg/día | 50 μg/kg/día | 70 μg/kg/día | 70 μg/kg/día | 70 μg/kg/día | 70 μg/kg/día | 90 μg/kg/día | 90 μg/kg/día |
| Puntuación total | 15 | 6 | 6 | 4 | 3 | 2 | 5 | 3 | 6 |
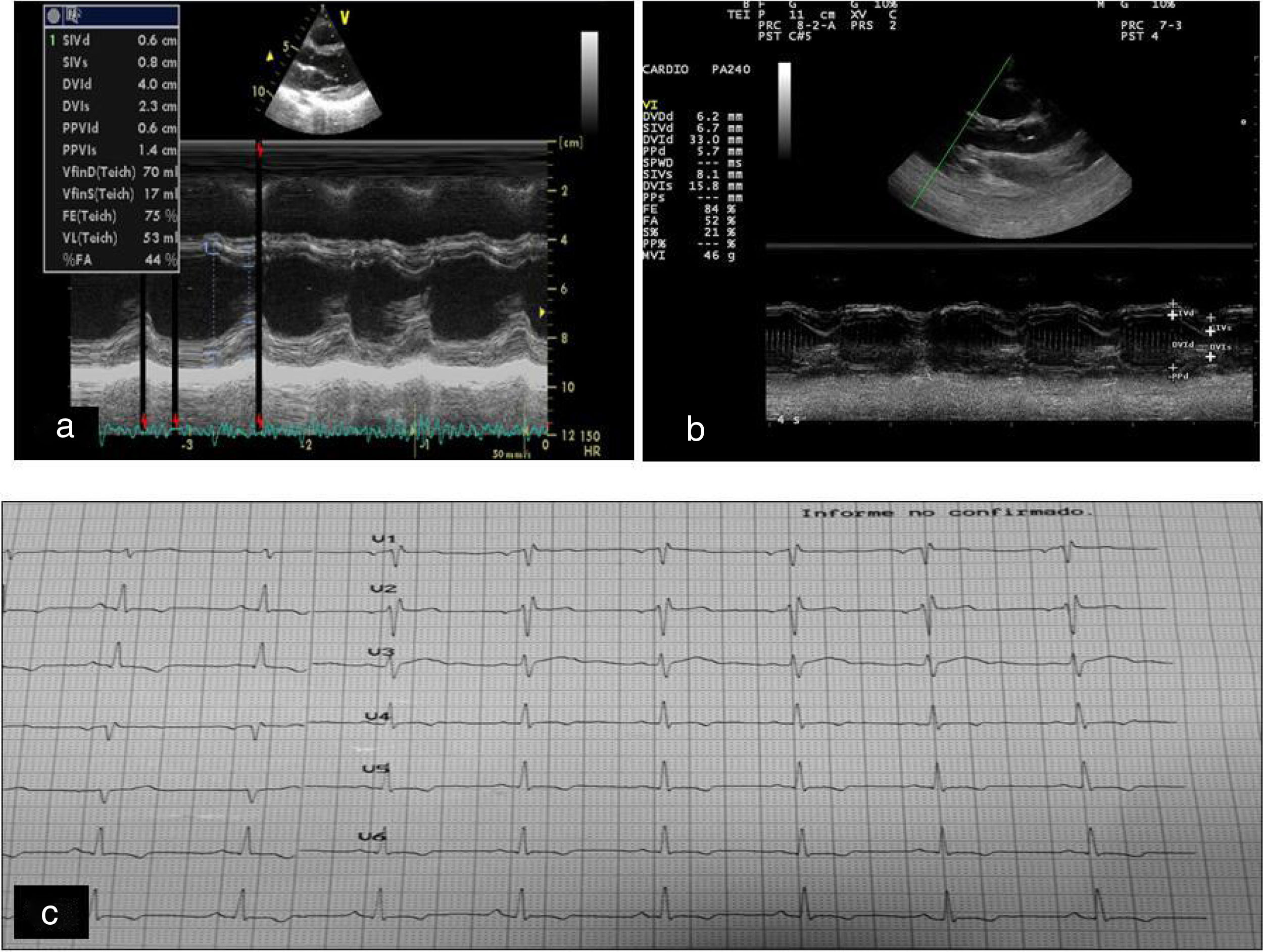
Desde el punto de vista cardiológico, se objetivó, por ecocardiografía, una discreta mejoría de la hipertrofia septal presente al inicio del estudio que se mantuvo estable hasta el final del mismo. El septo ventricular al final de la diástole disminuyó de 9mm en la situación basal a 7,2mm al año de imitar el tratamiento (fig. 1 a y b), sin disfunción ventricular sistólica o diastólica. La electrocardiografía basal reveló alteraciones inespecíficas en la repolarización con inversión de ondas T en precordiales izquierdas sin ondas Q patológicas (fig. 1 c), típicas de la ataxia cardíaca de Friedreich que persistieron hasta el final del estudio.
 Ecocardiografía basal. b) Ecocardiografía un año después del tratamiento con rhIGF-1: discreta reducción del septum interventricular y del diámetro diastólico del ventrículo izquierdo (DdVI). c) Cambios en la repolarización manifestados por cambios en las ondas T sin ondas q patológicas.")
a) Ecocardiografía basal. b) Ecocardiografía un año después del tratamiento con rhIGF-1: discreta reducción del septum interventricular y del diámetro diastólico del ventrículo izquierdo (DdVI). c) Cambios en la repolarización manifestados por cambios en las ondas T sin ondas q patológicas.
Respecto al perfil de seguridad, no se registraron eventos adversos a la terapia con rhIGF-1, con la excepción de reacciones inflamatorias menores locales en el punto de la inyección de 4-5 días de duración. La concentración sérica máxima de IGF-1 a dosis plenas de rhIGF-1 fue de 1071 ng/ml (valores de referencia, 180-1.000 ng / ml). El perfil glucémico permaneció normal durante todo el estudio.
En nuestro conocimiento, es el primer caso de AF tratado con rhIGF-1 en el que se consigue mejoría clínica sintomática y posterior estabilidad a los 3años de seguimiento. El tratamiento se inició al comienzo del deterioro neurológico y durante los primeros 3meses se objetivó el mayor cambio clínico en la situación neurológica de todo el estudio con un descenso de 9 puntos en la SARA. Es importante destacar que la mejoría clínica inicial progresó en los siguientes 12 meses y posteriormente se mantuvo estable, aunque con pequeñas oscilaciones en las puntuaciones de la SARA, que fueron contrarrestados con el aumento progresivo del rhIGF 1 hasta una dosis total de 90 μg/kg/día a los 36 meses. Su nueva situación neurológica le permitió mantener una independencia total para realizar actividades cotidianas como vestirse, comer, escribir, hacer ejercicio, etc., hasta el final del estudio.
Aunque el seguimiento de la afectación cardíaca no era un objetivo primario del estudio, se objetivó por ecocardiografía que el tratamiento con rhIGF-1 no solo no aumentó la hipertrofia cardíaca, sino que después del tratamiento documentamos una disminución del septo interventricular en telediástole. A pesar del interés de los datos obtenidos respecto al beneficio del tratamiento en la cardiopatía de nuestra paciente, los resultados necesitan ser interpretados con cautela principalmente por ser un caso aislado. Aunque parece que el tratamiento podría incrementar significativamente los niveles de frataxina en cardiomiocitos, al menos en un modelo de ratón transgénico (YG8R mice)21, se necesitan estudios con diseños adecuados y mayor tamaño muestral que puedan confirmar una utilidad real en estos pacientes.
El estudio refuerza la hipótesis del posible efecto neuroprotector que tiene el tratamiento con rhIGF-1 en ataxias cerebelosas degenerativas, como en el caso de los datos publicados por otro grupo español en un ensayo abierto con rhIGF-1 realizado en adultos con ataxias espinocerebelosas, en el que consiguieron estabilizar la progresión de la enfermedad (según la evaluación de SARA) durante al menos los 20 meses que duró el tratamiento16.
Aunque la efectividad del tratamiento con rhIGF-1 en pacientes con AF no está bien establecida, y puesto que nuestros hallazgos provienen del estudio de un caso clínico aislado, debemos señalar que pueden no ser aplicables a todos los casos de AF. Sin embargo, indica que la terapia con rhIGF-1 podría reducir o incluso mejorar el deterioro neurológico progresivo y detener/mejorar la miocardiopatía inherente a esta enfermedad.
Dado que actualmente no existe tratamiento curativo para la AF, creemos que cualquier fármaco potencialmente capaz de detener o retrasar la progresión de esta enfermedad sin producir efectos adversos graves merece una investigación adicional. Se requieren estudios controlados aleatorizados adicionales para confirmar los beneficios obtenidos en nuestro caso, intentar determinar el mecanismo de acción del rhIGF-1 y establecer lo que se puede esperar durante el tratamiento a largo plazo para esta y otras enfermedades neurodegenerativas.

