



El enanismo primordial osteodisplásico microcefálico (MOPD) es un síndrome caracterizado por la presencia de restricción del crecimiento intrauterino, deficiencia del crecimiento posnatal, microcefalia y un fenotipo similar al síndrome de Seckel1. El MOPD tipo ii es un síndrome raro, de herencia autosómica recesiva, y es el más distintivo dentro de este grupo de patologías; recientemente, nosotros describimos un paciente de origen colombiano, con una nueva mutación del gen PCNT en donde se presentaba un cambio en el exón 10, c. 1468C>T que resulta en la creación de un codón prematuro de parada en el aminoácido 490 p.Q490X, lo cual se predice genera una proteína truncada2.
Dicho paciente, en el momento del reporte tenía 5 años de edad, se caracterizaba por presentar retardo en el desarrollo psicomotor, talla extremadamente baja (por debajo del percentil 3 para la edad), microcefalia, fisuras palpebrales inclinadas hacia abajo, nariz prominente, amelogénesis imperfecta, desviación ulnar, pelvis alta y estrecha, piernas y brazos desproporcionalmente cortos, coxa vara, voz aguda, y personalidad social2. Entre los estudios de extensión solicitados a los 5 años de edad se incluían una tomografía computarizada (TC) simple de cerebro en la cual se identificó un iv ventrículo y una fosa posterior normales, con cierre de las suturas metópica y sagital, y permeabilidad de la sutura coronal y lambdoidea; y una angiografía, la cual fue negativa para aneurismas y enfermedad de moyamoya; se solicitó prueba molecular que confirmaba diagnóstico clínico.
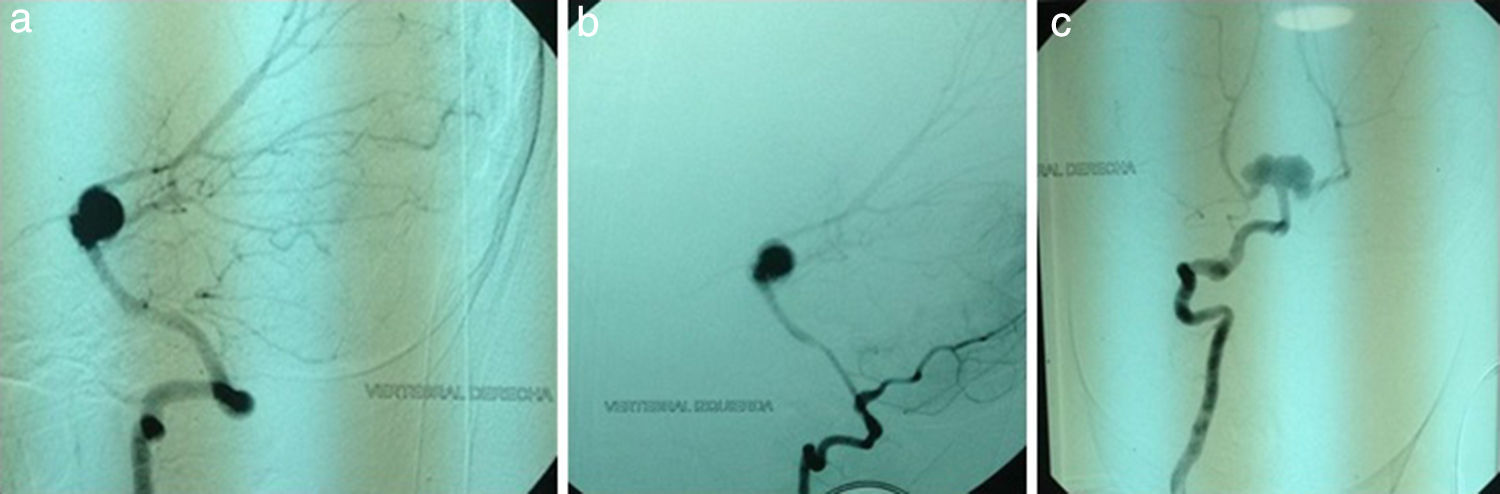
Actualmente, con 7 años de edad, el paciente presenta episodio de cefalea súbita asociada a náuseas, vómito, convulsiones, pérdida de conocimiento y déficit motor. Se realiza al paciente una TC cerebral, en la cual se observa hemorragia subaracnoidea; se complementan estudios con angiografía, que reporta a nivel de carótida interna izquierda una dilatación infundibular en el segmento de la comunicante posterior; aneurisma de 3mm de tipo transicional en la carótida izquierda; en la carótida interna derecha se observa una dilatación infundibular en el segmento de la comunicante posterior y aneurisma del tope de la basilar de aproximadamente 13mm, con signos claros de rotura trilobulada que compromete el nacimiento de ambas cerebrales posteriores (fig. 1 A-C). El paciente es tratado mediante una intervención endovascular, posterior a la cual el paciente presentó leve mejoría de su estado de consciencia, pero persiste con cuadriparesia y dificultad severa para el habla.
 Arteria vertebral derecha. b) Arteria vertebral izquierda. c) Arteria vertebral derecha y basilar. Nótese aneurisma del tope de la basilar de aproximadamente 13 mm, que compromete el nacimiento de ambas cerebrales posteriores.")
La malformación de moyamoya y la aparición de múltiples aneurismas han sido ampliamente asociadas con diferentes síndromes genéticos, particularmente con MOPD tipo ii. Hall et al. (2004) documentaron en un grupo de 58 pacientes la presencia de aneurismas o moyamoya en el 19,0% (11 pacientes)3. Brancati et al. (2005) documentaron los mismos eventos en 15 de 63 pacientes (23,8%)4 y Bober et al. (2010) en 13 de 25 (52%)5. En dichas publicaciones se evidencia un predominio en el género masculino para la aparición de enfermedad cerebrovascular3-5, sin embargo, persiste la inquietud de si el género femenino ejerce un efecto protector o si el hecho de pertenecer al sexo masculino aumenta el riesgo para dicha alteración5.
El compromiso neurológico secundario a las alteraciones cerebrovasculares presenta un espectro amplio secuelas que pueden variar desde disfasia residual hasta la muerte súbita3-5. En la cohorte de Brancati et al. (2005) se evidenció que los pacientes con enfermedad de moyamoya presentan complicaciones cerebrovasculares a menos edad; sin embargo, aquellos con aneurismas intracraneales de aparición posterior presentaban un peor pronóstico debido a hipertensión crónica severa y miocardiopatía dilatada4.
El diagnóstico de aneurismas múltiples y/o enfermedad de moyamoya es particularmente difícil en pacientes con MOPD tipo ii, debido a las dificultades asociadas a las alteraciones anatómicas propias de la patología, como lo es un sistema vascular particularmente pequeño y arterias anormalmente tortuosas6. Debido a la estrecha relación que presenta el síndrome de MOPD tipo ii con la aparición de patologías cerebrovasculares, es importante resaltar con la actualización de este caso clínico, la relevancia de realizar en estos pacientes tamizaje anual desde el momento del diagnóstico5, mediante resonancia magnética o angiografía con el objetivo de prevenir eventos potencialmente discapacitantes y/o posiblemente mortales5, permitiendo un abordaje temprano que disminuya complicaciones quirúrgicas, permitiendo una mayor calidad de vida y supervivencia a los pacientes. De igual manera, nosotros consideramos que aquellos pacientes con clínica indicativa de síndrome de MOPD o confirmada por estudios genéticos deben ser evaluados mediante angiorresonancia de manera rutinaria para descartar aneurismas y/o malformación de moyamoya.
FinanciaciónUniversidad Icesi.
Centro de Investigaciones en Anomalías Congénitas y Enfermedades Raras (CIACER).
Conflicto de interesesNinguno por declarar.

