



La enfermedad de Creutzfeldt-Jakob (ECJ) es la forma más común de enfermedad priónica en humanos, resultado de un cambio conformacional en la proteína priónica (PrP) normal (PrPC) en una forma patológica (PrPSc), con agregados en el sistema nervioso central1. La ECJ puede clasificarse etiológicamente en infecciosa/transmitida, esporádica (ECJe) o familiar (ECJf)2. Las formas familiares de la ECJ son causadas por mutaciones en la codificación de genes de la PrP; el grupo más grande de ECJf existe en judíos de ascendencia libia y tunecina portadores de la mutación E200K (sustitución de Glu por Lys) en el gen PRNP3,4. Entre los años 1997 y 2008, un centro de referencia en ECJ estableció que de 517 pacientes analizados un 15,6% presentó la mutación E200K5. El diagnóstico definitivo de ECJ se basa en la identificación histológica de degeneración espongiforme en tejidos del sistema nervioso central. El diagnóstico probable se basa en los hallazgos clínicos, los patrones típicos del electroencefalograma (EEG) y/o la aparición de la proteína 14.3.3 en el líquido cefalorraquídeo junto a menos de 2 años de duración de la enfermedad2. En fases precoces de la enfermedad, el EEG puede ser normal, o mostrar cambios inespecíficos, como actividades delta o theta. Ocasionalmente, el primer hallazgo puede ser la aparición de actividades delta rítmicas intermitentes frontales (FIRDA) o descargas epileptiformes lateralizadas periódicas (PLED). Posteriormente, en el plazo de pocas semanas, pueden aparecer complejos periódicos de ondas agudas (PSWC) con una frecuencia de 1-2 Hz6. En pacientes con la mutación E200K las alteraciones del sueño constituyen una forma frecuente de presentación, mostrando en la videopolisomnografía (v-PSG) una arquitectura anormal del sueño, múltiples sacudidas y movimientos7, así como apneas centrales y obstructivas1.
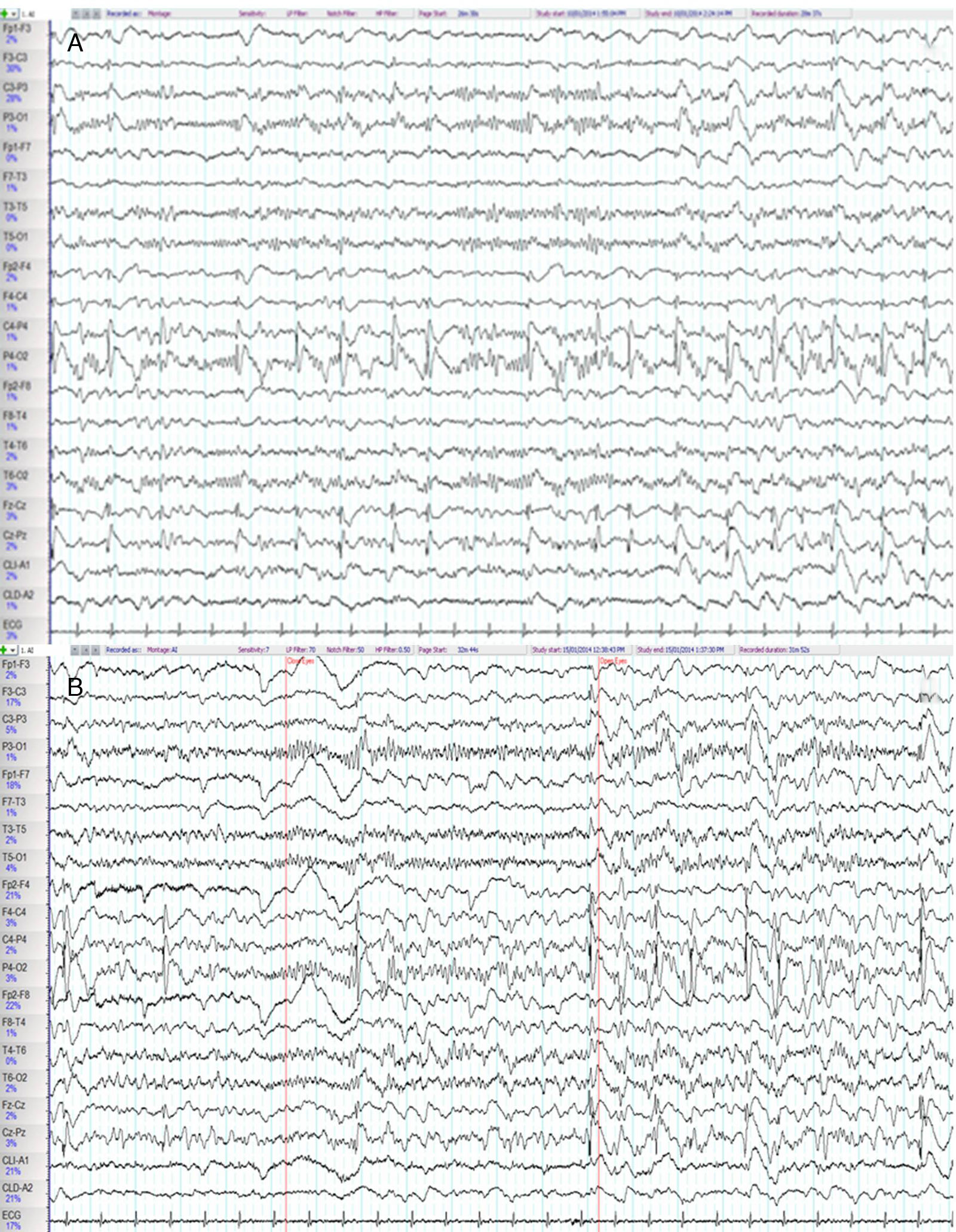
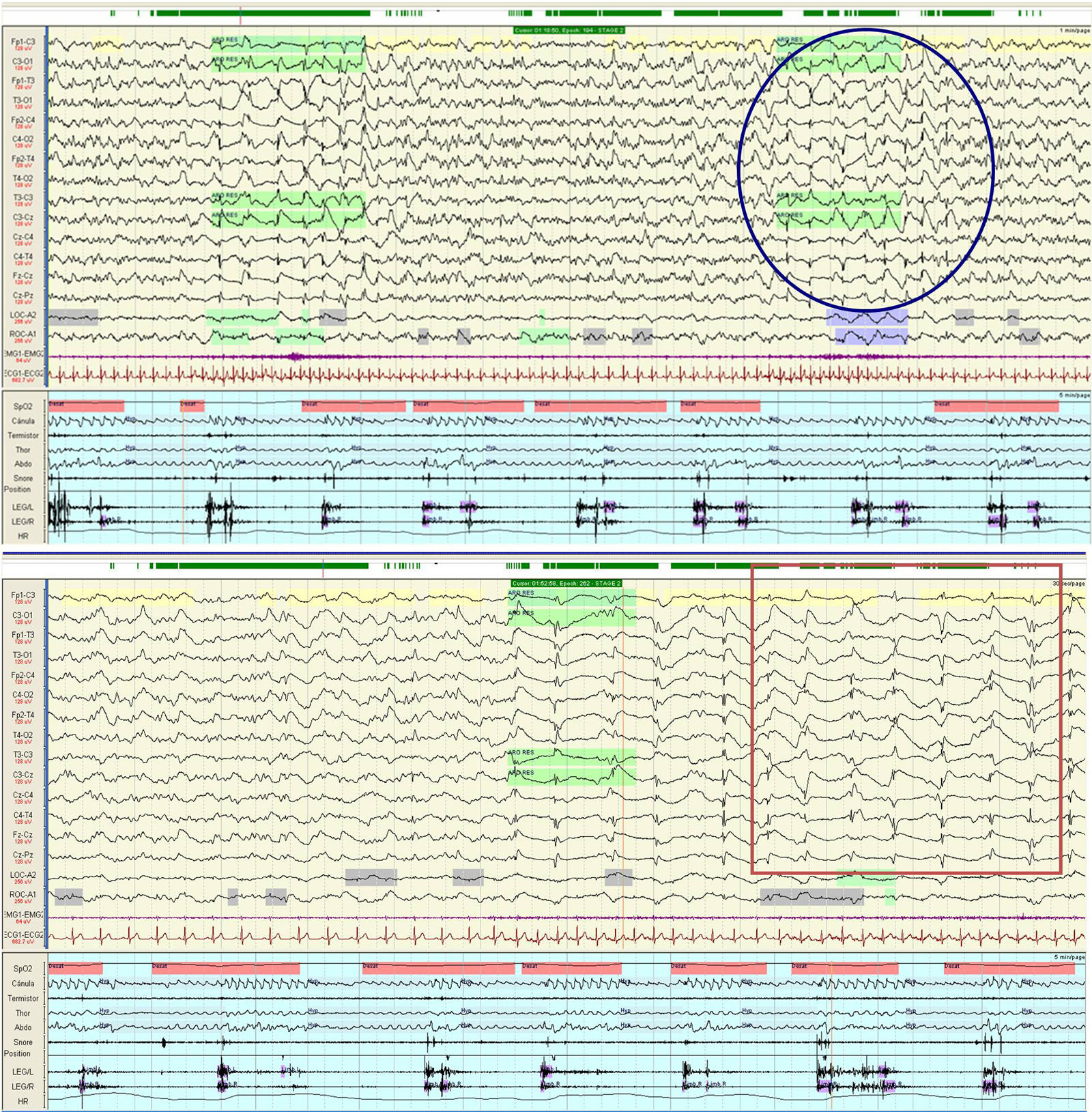
Presentamos el caso de un varón de 53 años, con antecedentes personales de síndrome de apnea hipopnea del sueño y antecedentes familiares de padre fallecido 11 años antes por ECJ. Ingresó por presentar un cuadro de unas 10 semanas de evolución caracterizado por progresiva dificultad para la emisión del lenguaje, así como inestabilidad de la marcha y movimientos anormales en el miembro superior izquierdo. En la exploración presentaba alteración del lenguaje, espasticidad generalizada, movimientos distónicos en la mano izquierda, mioclonías generalizadas, ataxia de la marcha y signos de piramidalismo. En la resonancia magnética cerebral (RM) se evidenció una restricción a la difusión y un aumento de señal en cuerpo estriado bilateral, corteza insular, cíngulo y áreas frontales de ambos hemisferios, predominando en el hemisferio derecho. Se realizaron 2 EEG, el día 2 y 7 tras el ingreso, mostrando una actividad epileptiforme constituida por complejos punta-onda a 1Hz sobre la región parietal derecha y línea media, que en ocasiones se acompañaba de mioclonías faciales y sacudidas clónicas de la mano izquierda, manteniendo un ritmo alfa posterior reactivo (fig. 1). El estudio v-PSG que se realizó a la semana del ingreso mostró por primera vez en vigilia y en los arousals una actividad periódica generalizada de ondas bi y trifásicas a 1-2Hz, así como una ausencia de los grafoelementos de cada una de las fases del sueño NREM y REM, presentando un patrón alternante cíclico (PAC). Además, a pesar del uso adecuado de la presión positiva continua en la vía aérea durante el registro, se evidenció un índice de apnea hipopnea mayor de 50 eventos/h (fig. 2). El paciente permaneció ingresado en el Servicio de Neurología, progresando hasta el mutismo acinético, falleciendo tras 2 meses de ingreso hospitalario. El estudio genético evidenció la presencia de la mutación p.Glu200Lys (p.E200K), en la posición 598 del exón 2 del gen PRNP del paciente, confirmando el diagnóstico de ECJf.
 Se observan sobre la región parietal derecha y línea media (C4-P4, P4-O2, Fz-Cz, Cz-Pz) frecuentes descargas de actividad epileptiforme constituidas por complejos punta-onda a 1Hz, que en ocasiones difunden a regiones vecinas. B) Sobre las regiones posteriores (P3-O1, T5-O1, P4-O2, T6-O2) se registra un ritmo alfa parieto-occipital, reactivo (que aparece al cerrar los ojos y se atenúa al abrirlos).")
Electroencefalograma inicial. A) Se observan sobre la región parietal derecha y línea media (C4-P4, P4-O2, Fz-Cz, Cz-Pz) frecuentes descargas de actividad epileptiforme constituidas por complejos punta-onda a 1Hz, que en ocasiones difunden a regiones vecinas. B) Sobre las regiones posteriores (P3-O1, T5-O1, P4-O2, T6-O2) se registra un ritmo alfa parieto-occipital, reactivo (que aparece al cerrar los ojos y se atenúa al abrirlos).
 correspondiente a un arousal postevento respiratorio, donde se observa una actividad generalizada de ondas bi o trifásicas a 1Hz asociada a un aumento de la frecuencia cardiaca. El cuadrado señala la fase 2 del PAC, caracterizada por ondas lentas difusas, destacando la ausencia de grafoelementos típicos de las distintas fases del sueño.")
Videopolisomnografía nocturna. Se observa dentro del círculo la fase 1 del patrón alternante cíclico (PAC) correspondiente a un arousal postevento respiratorio, donde se observa una actividad generalizada de ondas bi o trifásicas a 1Hz asociada a un aumento de la frecuencia cardiaca. El cuadrado señala la fase 2 del PAC, caracterizada por ondas lentas difusas, destacando la ausencia de grafoelementos típicos de las distintas fases del sueño.
Los hallazgos del EEG se relacionan con los publicados en la literatura, en los que en fases precoces de la ECJ aún puede mantenerse un ritmo posterior reactivo. Las actividades FIRDA o los PLED pueden ser los predecesores de la PSWC8. Appel et al.9 encontraron en pacientes con la mutación E200K correlación entre la actividad periódica y la afectación cortical en RM. De acuerdo con otros estudios10, la incidencia de crisis es menor en pacientes con la mutación E200K respecto a la ECJ esporádica, debido a la afectación de regiones menos epileptógenas de la sustancia gris profunda9. Al igual que en estudios previos, el estudio v-PSG muestra la existencia de un PAC11. Cohen et al.1 encontraron patología severa del sueño en pacientes con mutación E200K; estos hallazgos aparecieron precozmente en el curso de la enfermedad, mostrando en el estudio v-PSG desaparición de los husos del sueño, muy baja eficiencia del sueño y ausencia de sueño REM1. En un reciente estudio sobre 28 pacientes consecutivos con autopsia de ECJ, un 90% de ellos presentaron anomalías del sueño en el momento de su evaluación inicial7. La rotura en la arquitectura del sueño y la desaparición de los husos de sueño se han relacionado con la participación de estructuras talámicas1. De acuerdo con la literatura, consideramos que serán precisos más estudios para valorar el papel del sueño en el diagnóstico precoz de pacientes con sospecha de ECJ, especialmente en las formas familiares.

