



El análisis genético de los trastornos específicos del lenguaje resulta del máximo interés, tanto para la práctica clínica como para la teoría lingüística. No obstante, un resultado casi universal de dicho análisis es que no parece existir una relación unívoca y obligada entre la mutación de determinados genes y la aparición de patologías concretas al término del desarrollo, ni por consiguiente, una relación causal directa entre el genotipo y el fenotipo.
ObjetivosEl presente trabajo se plantea evaluar esta clase de evidencias (utilizando como modelo, allí donde resulte ilustrativo, el gen FOXP2, considerado habitualmente como el «gen del lenguaje» por excelencia), proponer posibles causas que expliquen su recurrencia y discutir si tales explicaciones contribuyen realmente a esclarecer la genuina etiología de estos trastornos.
ResultadosLa clave para entender el intrincado (y a primera vista desconcertante) escenario resultante del análisis genético de los trastornos específicos del lenguaje radica en atender al verdadero papel que desempeñan los genes durante la ontogenia y, especialmente, al modo en que se regulan los procesos de desarrollo: lejos de erigirse en los agentes causales directos responsables en exclusividad de la aparición de los fenotipos, los genes constituyen uno más de los múltiples factores implicados.
ConclusionesLa asunción de la complejidad de dicho papel, así como la conveniencia de considerar modelos alternativos del desarrollo, menos centrados en los genes (por paradójico que pueda parecer), permite explicar satisfactoriamente el modo en que las alteraciones génicas contribuyen a la aparición de este tipo de trastornos de la cognición.
Genetic analysis of specific language disorders is of major interest for both clinical research and linguistic theory. However, the results of this analysis almost always do not show any univocal and compulsory relationships between particular gene mutations and particular disorders or a casual link between the genotype and the phenotype.
ObjectivesThis paper will review this type of evidence (referring to the «language gene» FOXP2 as a leading example, where possible), try to suggest plausible reasons for such a perplexing output, and ultimately discuss if such reasons really explain the genuine aetiology of these conditions.
ResultsThe key to disentangle and understand the puzzling scenario emerging from the genetic analysis of specific language disorders is to pay attention to the actual role played by genes during ontogeny and, in particular, to the way in which developmental processes are actually regulated: genes are not direct causal agents regarding the emergence of impaired or wild phenotypes, but just one among the diverse types of regulatory factors involved.
ConclusionsWhen such a complex role as well as development models less focused on the genes are considered, the way in which genetic mutations really contribute to the emergence of these cognitive disorders is quite satisfactorily explained.
La posibilidad de identificar y determinar la naturaleza estructural y funcional de genes relacionados con el lenguaje (es decir, de genes cuyos productos desempeñarían presumiblemente un papel relevante en la regulación del desarrollo y el funcionamiento de los centros neuronales que intervienen en el procesamiento de estímulos lingüísticos) resulta del máximo interés por dos razones fundamentales. Por una parte, porque podría servir para corroborar determinadas hipótesis surgidas en el campo de la Lingüística que sostienen que la competencia gramatical adquirida por el individuo al término de su desarrollo (esto es, el conocimiento que alcanza acerca de su propia lengua) no podría explicarse únicamente como el resultado de un proceso de aprendizaje de carácter inductivo a partir de los datos que integran el input lingüístico al que se ve expuesto1,2. De hecho, y siguiendo al propio Chomsky1, quienes defienden este tipo de hipótesis innatistas han llegado a postular la existencia de un genotipo lingüístico que consistiría en toda aquella información no derivable de la experiencia que es imprescindible para la adquisición del lenguaje, en esencia, la necesaria para la construcción de la Gramática Universal3. Así, el lenguaje se adquiriría porque el input lingüístico promovería la aparición del fenotipo que representa la competencia a partir de dicho genotipo3. Por otro lado, la existencia de numerosos trastornos del lenguaje que poseen un carácter hereditario4–6 parece sugerir que el esclarecimiento de su genuina etiología pasa necesariamente por la identificación y la caracterización de las mutaciones que han debido afectar presumiblemente a alguno de los genes que integran dicho genotipo. A este respecto, una hipótesis de trabajo habitual en el área es que la presencia en determinados sujetos de un patrón de actuación lingüística anómalo de carácter hereditario debe ser el resultado de la mutación de genes específicos que, afectando a un determinado componente de la competencia, deja inalteradas las restantes capacidades cognitivas (algunas de las cuales estarán implicadas en la actuación). En último término, ello será posible porque la mutación de dichos genes da lugar a alteraciones estructurales y/o funcionales en áreas cerebrales implicadas en el procesamiento del lenguaje.
En los últimos años se han identificado y caracterizado clínicamente diversos síndromes, afecciones, trastornos o enfermedades de carácter hereditario en las que sólo el lenguaje parece estar afectado. Entre otros cabe mencionar el trastorno específico del lenguaje (en adelante, TEL) (OMIM 602081), la dislexia (OMIM 127700), el trastorno de los sonidos del habla (en adelante, SSD) (OMIM 608445) y posiblemente algunos trastornos adicionales de prevalencia mucho menor, entre los que podrían encontrarse el síndrome de Landau-Kleffner (OMIM 245570), la epilepsia rolándica (o silviana) con dispraxia verbal (OMIM 601085) o el síndrome de la deleción del fragmento 22q13.3 (OMIM 606232). Asimismo, se han identificado diversos genes que cabe considerar como candidatos o como factores de riesgo para su aparición, así como distintos loci (esto es, lugares físicos en el cromosoma) ligados o asociados a dichos trastornos4–6. Sin embargo, y por paradójico que pudiera resultar en una primera instancia, lo cierto es que el análisis genético de este tipo de trastornos no ha contribuido necesariamente a necesariamente su genuina etiología, toda vez que parece sugerir que las relaciones existentes entre el genotipo y el fenotipo no son tan directas como cabría imaginar.
En el presente trabajo se discuten algunas de las evidencias en este sentido, ilustrándolas fundamentalmente con datos derivados del análisis del gen FOXP2, considerado habitualmente como uno de los factores causales del trastorno específico del lenguaje y al mismo tiempo como «el gen del lenguaje» por excelencia7–12. A la luz se dichas evidencias, se discutirá la necesidad de reexaminar las relaciones existentes entre el genotipo y el fenotipo si se quiere entender el verdadero papel que desempeñan los genes en la aparición de estos trastornos del neurodesarrollo, lo que obligará, en último término, a abandonar concepciones excesivamente gen-céntricas de la ontogenia cerebral.
DesarrolloAlgunos resultados aparentemente problemáticos del análisis genético de los trastornos específicos del lenguaje: FOXP2 como paradigmaEl escenario resultante del análisis de los genes ligados o asociados a los diversos trastornos específicos del lenguaje resulta particularmente intrigante:
- –
En general, el grado de afectación de los individuos que presentan una misma variante anómala de cualquiera de estos genes candidato es diverso (penetrancia variable), sucediendo en ocasiones que el trastorno no llega a manifestarse (penetrancia nula) o lo hace de manera muy leve (penetrancia reducida). Así, los individuos que portan la mutación R553H del gen FOXP2 (la primera en ser identificada) presentan síntomas hasta cierto punto diferentes13,14, mientras que recientemente se ha descrito una mutación por completo asintomática (T1591C) en determinados miembros de un mismo linaje15.
- –
Habitualmente se observa que diferentes mutaciones de un mismo gen originan fenotipos (ligeramente) distintos, que en ocasiones son susceptibles de caracterizarse clínicamente como trastornos diferentes, los cuales pueden ser específicamente lingüísticos, pero también revestir un carácter cognitivo general, afectando simultáneamente al lenguaje y a otras capacidades cognitivas (en otras circunstancias se observa sencillamente que un mismo locus está ligado o asociado a fenotipos o trastornos diferentes). Así, en el caso particular de FOXP2, el trastorno asociado a la mayor parte de las diferentes mutaciones puntuales descritas hasta el momento (y a la mayoría de los eventos de translocación cromosómica que afectan a la secuencia del gen) se ha descrito como una dispraxia orofacial ligada al desarrollo, o bien como una disartria espástica, aunque su categorización clínica precisa y la naturaleza exacta del déficit subyacente (¿un trastorno del habla?; ¿un trastorno del lenguaje?; ¿un trastorno cognitivo general que incluye déficits lingüísticos?; ¿un trastorno motor que afecta al lenguaje?) sigue siendo objeto de controversia9,13,14,16. En todo caso, la última de las mutaciones en ser identificada (T1591C) da lugar a un fenotipo sustancialmente diferente y más complejo, que incluye episodios epilépticos focalizados, un déficit cognitivo y diversos déficits lingüísticos15. En último extremo, determinados polimorfismos del gen se han asociado con trastornos cognitivos netamente distintos, como la esquizofrenia17 o la demencia lobular frontotemporal18, aunque sin llegar a considerarlos factores causales o de riesgo en su aparición. Parece evidente que diferentes mutaciones de FOXP2 dan lugar a procesos de desarrollo ligeramente distintos de las áreas cerebrales en las que se expresa el gen, y que las alteraciones neurológicas resultantes de dichas mutaciones pueden condicionar la intensidad y/o el modo con que se manifiestan clínicamente multitud de trastornos cognitivos, plausiblemente a través de su efecto sobre sus endofenotipos de carácter lingüístico, como pone paradigmáticamente de manifiesto el efecto modulador que los polimorfismos rs1456031 TT y rs17137124 TT ejercen en la presentación de la demencia lobular frontotemporal merced a su influencia sobre la fluidez verbal18. Y todo ello dejando al margen la circunstancia de que dichas áreas difícilmente pueden considerarse como dedicadas exclusivamente al procesamiento de estímulos de carácter lingüístico, como también el hecho de que su alteración, causada por la mutación de otros genes diferentes, suele dar lugar a déficits y trastornos de distinta naturaleza, incluidos los de índole no lingüística. Así, la patología primaria asociada a la mutación del gen FOXP2 parece localizarse en los ganglios basales14,19, cuyas funciones, de índole motora y cognitiva, trascienden, sin embargo, las relacionadas específicamente con el lenguaje20,21. Al mismo tiempo, la disfunción de esta estructura subcortical, debida a la mutación de otros genes distintos de FOXP2, origina patologías, no exclusivamente lingüísticas, como la enfermedad de Huntington22, la enfermedad de Parkinson20,23, la 3-α-metilglutaconicaciduria24, la glutaricacidemia de tipo I25 o la parálisis supranuclear progresiva26,27.
- –
Por otro lado, conviene tener presente que los productos codificados por buena parte de los genes cuya mutación da lugar a trastornos de esta índole no actúan de forma aislada, sino que para ser funcionales deben integrarse en complejos de carácter multiproteínico. En el caso concreto de la proteína FOXP2, la especificidad de su unión al ADN (en términos temporales y/o tisulares) parece venir condicionada y/o facilitada por su interacción con otras moléculas de FOXP2 (homodimerización), pero también merced a la formación de heterodímeros con FOXP1 o FOXP4, y a su unión con el co-represor transcripcional CtBP128,29. Significativamente, la mutación de los genes que codifican estas proteínas parece dar lugar también a alteraciones del lenguaje y del habla, como ejemplifica el caso de FOXP130–32. No obstante, y como también pone de manifiesto lo que sucede con este gen, los déficits observados a este nivel no recapitulan con exactitud los asociados a la mutación del gen FOXP2 (así, en la mayoría de los casos la mutación de FOXP1 no causa una dispraxia verbal30,33), mientras que es frecuente la aparición simultánea de trastornos cognitivos de diversa índole (así, la mutación de FOXP1 se ha asociado con un retraso mental32 y con el autismo31).
- –
Es preciso tener en cuenta, asimismo, que los productos codificados por la mayor parte de estos genes actúan habitualmente integrados en complejas redes reguladoras5. Sucede, sin embargo, que la mutación de algunas de las dianas de estos genes no origina un fenotipo semejante al causado por la mutación del gen regulador, sino que puede dar lugar a síntomas susceptibles de ser caracterizados clínicamente como indicativos de la existencia de un trastorno diferente. Es probablemente a este nivel, y en relación con la red reguladora de la que forma parte específicamente FOXP2, donde se están produciendo actualmente los avances más significativos en la caracterización de las bases genéticas del lenguaje. La reciente dilucidación de una fracción de los componentes que integran dicha red no ha hecho sino confirmar el importante papel que desempeña el gen en la modulación del desarrollo de los centros cerebrales implicados en el procesamiento lingüístico34–36. Una de las dianas de FOXP2 es CNTNAP2, que codifica una proteína de la familia de las neurexinas que parece intervenir en la regulación de la sinaptogénesis37, participando específicamente en el establecimiento del patrón de interconexión privativo del lóbulo frontal38. No obstante, el gen aparece mutado en individuos afectados por diversos trastornos neurológicos, incluido el TEL39, diferentes variantes de retrasos o trastornos lingüísticos40,41 o el autismo42,43. Otra de las dianas de FOXP2ha resultado ser SRPX2, que codifica una proteína con tres motivos «sushi» consenso y un motivo HYR, la cual podría estar implicada en la maduración perinatal y posnatal de determinados circuitos del córtex cerebral (incluidos los que intervienen en el control del habla), teniendo en cuenta el papel que desempeña en la regulación de la migración y la adhesión celulares44,45. El gen SRPX2 se venía considerando el candidato principal para una variante de epilepsia rolándica (o silviana) con dispraxia verbal similar a la descrita por Scheffer et al46, aunque ligada al cromosoma X, la cual se caracteriza, entre otros síntomas, por la presencia de una apraxia oromotora y dificultades leves para la comprensión de determinadas estructuras lingüísticas44. Sin embargo, la mutación del gen parece ser la causante, asimismo, de una variante de polimicrogiria perisilviana bilateral44, un trastorno entre cuyos síntomas distintivos se encuentran la disartria (o en ciertos casos la ausencia de lenguaje hablado), así como un leve retraso mental47. Se ha sugerido que la epilepsia rolándica (o silviana) con dispraxia verbal, y en general, las epilepsias infantiles benignas con picos centrotemporales (o epilepsias rolándicas), podrían representar el extremo de un continuo fenotípico del que también formaría parte el denominado síndrome de descargas continuas en pico y onda durante la fase de sueño lento (CSWS)44, y cuyo otro extremo correspondería al síndrome de Landau-Kleffner46. El endofenotipo más característico de todos estos trastornos, a saber, el denominado CTS (picos centrotemporales nítidos focales difásicos) se ha asociado con el gen ELP448. Este gen codifica uno de los componentes del denominado complejo elongador, un complejo multiproteínico con actividad histona acetilasa implicado en la transcripción del ARN y la modificación de los ARNt, cuya disfunción se ha correlacionado con una alteración de la movilidad y la migración celulares, que afectaría, en particular, a determinadas poblaciones neuronales durante el desarrollo del córtex cerebral49. Significativamente, el locus correspondiente al gen ELP4 se ha relacionado, asimismo, con el SSD50, lo que sugiere que la comorbilidad observada entre determinadas formas de epilepsia rolándica y el SSD podría deberse a un efecto pleiotrópico de este gen. Por lo demás, una vez segregada, la proteína SRPX2 interactúa con diversas proteínas, en particular, con PLAUR, que codifica el receptor del activador del plasminógeno51. Sucede que tanto SRPX2 como PLAUR son dianas de FOXP215, con la particularidad de que la variante R553H del gen es incapaz in vitro de reprimir la expresión de ambos genes15. En resumen, distintos genes que parecen formar parte de una misma red reguladora (probablemente FOXP2, CNTNAP2, SRPX2, PLAUR y ELP4) se encuentran relacionados con trastornos sustancialmente diferentes en términos clínicos (posiblemente TEL, SSD, autismo, dispraxia, apraxia, epilepsias rolándicas, CSWS y síndrome de Landau-Kleffner), dando lugar su mutación a síntomas y endofenotipos diversos, tanto lingüísticos como no lingüísticos (déficits lingüísticos, déficits cognitivos, déficits oromotores, CTS).
- –
Adicionalmente, para cada uno de los trastornos específicos del lenguaje se han propuesto diversos genes candidato y múltiples genes que cabe considerar como factores de riesgo para su aparición, en esencia, porque la identidad de dichos genes difiere (hasta cierto punto) de una población a otra y/o dependiendo del subtipo de que se trate. Así, el TEL puede estar causado por la mutación del gen FOXP2 (aunque esta etiología es objeto de controversia, puesto que su caracterización como un trastorno sensorimotor14,16 sería incompatible con los criterios diagnósticos de esta patología52,53), si bien se describe mejor como el resultado del efecto acumulativo de diversos genes de menor importancia54. Hasta el momento se han identificado distintos QTL ligados o asociados al TEL55,56 y se han propuesto diversos genes candidato para el trastorno. Dejando al margen el caso ya discutido de CNTNAP2, existirían otros dos cuyos productos estarían implicados en la regulación del metabolismo del calcio, como son ATP13A4, que codifica una ATPasa transportadora de cationes de tipo P557 y ATP2C2, que codifica un transportador implicado en la translocación de iones calcio y manganeso desde el citosol al aparato de Golgi58. Un último candidato sería CMIP, que codifica un componente del dispositivo molecular de anclaje de la membrana al citoesqueleto celular y que podría intervenir en la regulación de la migración neuronal y/o la formación de los complejos sinápticos59. Significativamente, tanto ATP2C2 como CMIP se han asociado con el componente fonológico de la memoria de trabajo a corto plazo60, que constituye uno de los endofenotipos característicos (y probablemente el déficit nuclear) no sólo del TEL61, sino también de la dislexia62 y el SSD63. Aunque hasta el momento no ha sido posible relacionar ninguno de estos genes con la red reguladora de la que forma parte FOXP2, se sabe que CMIP se une in vitro a FLNA, una fosfoproteína que podría intervenir en la regulación de la migración neuronal durante la embriogénesis64 y que está codificada por un gen cuya mutación origina en ciertos casos trastornos del desarrollo que afectan especialmente al lenguaje65.
- –
Finalmente, sucede con frecuencia que existen individuos afectados por cualquiera de estos trastornos en los que la secuencia de los genes candidato es normal (fenocopia).
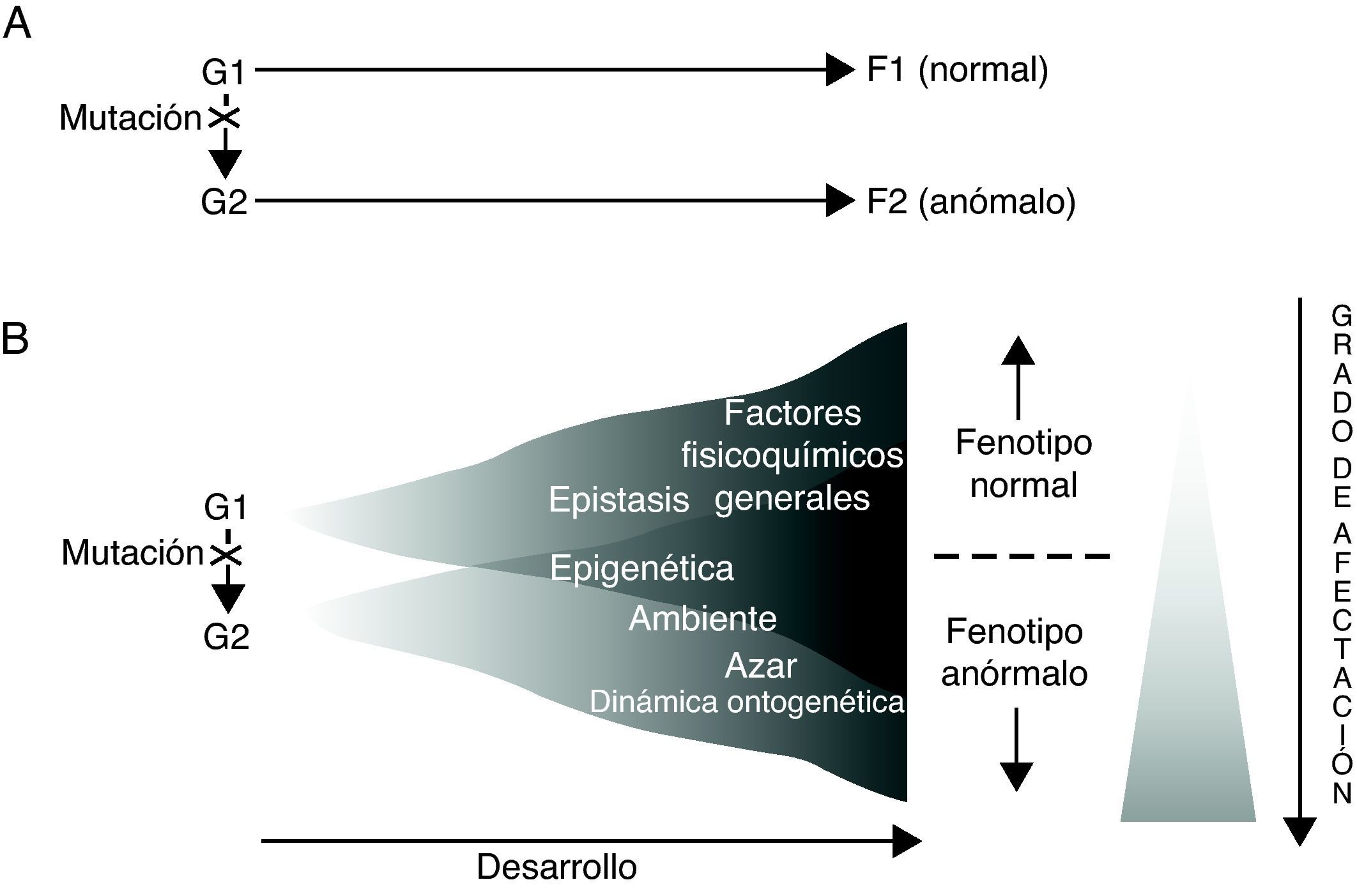
Los resultados derivados del análisis genético de los trastornos específicos del lenguaje, tal como se caracterizaron en el apartado anterior, parecen sugerir, cuando menos, dos cosas. Por un lado, que para cada trastorno existen necesariamente numerosos genes que cabe considerar como factores desencadenantes o de riesgo para su aparición (poligenismo), mientras que simultáneamente cada uno de esos genes desempeñará funciones diferentes en momentos y lugares distintos del organismo durante el desarrollo (pleiotropismo) (así, por ejemplo, durante el desarrollo embrionario FOXP2 no sólo se expresa en el cerebro, sino que también lo hace en el pulmón, el intestino y el corazón66). Por otro lado, que no existe (ni puede existir) una relación causal directa entre el genotipo y el fenotipo, y ello por dos razones fundamentales: en primer lugar, debido a la genuina naturaleza de los genes y al modo en que ejercen su actividad; en segundo lugar, a causa de la intervención de otros muchos elementos de naturaleza no genética en la regulación del desarrollo (fig. 1). En realidad, los genes no constituyen la causa primera de los procesos ontogenéticos (normales o anómalos), sino uno más de entre los múltiples factores involucrados, los cuales actuarán siempre en términos de paridad, en el sentido de que todos serían igualmente indispensables para la consecución del fenotipo final67–69, teniendo en cuenta además que el desarrollo es siempre el resultado de una interacción sinérgica y no meramente aditiva70.
-fenotipo (F) en relación con los trastornos del lenguaje. Adaptado de Sholtis y Weiss72.")
Dos modos alternativos de concebir las relaciones genotipo (G)-fenotipo (F) en relación con los trastornos del lenguaje. Adaptado de Sholtis y Weiss72.
Diversas razones parecen justificar lo anterior:
- –
Para empezar, resultaría erróneo considerar los genes como secuencias de ADN que contienen per se la información necesaria y suficiente para la síntesis de una determinada proteína funcional (o de determinados productos bioquímicos, puesto que no todos los genes codifican proteínas71). Antes bien, la mayoría de los genes es objeto de una modificación postranscripcional, de modo que pueden generarse proteínas alternativas (y funcionalmente diferentes) a partir de un mismo transcrito primario72, o incluso de genes diferentes, cuando se forman transcritos quiméricos73. Adicionalmente, para ser funcionales muchas proteínas deben modificarse postraduccionalmente (esto es, una vez sintetizadas) y/o integrarse en complejos multiproteínicos mediante su asociación con otras proteínas diferentes. Pero además deben actuar en los lugares celulares idóneos, lo que exige su transporte y su translocación merced a diversos mecanismos reguladores del tráfico intracelular de productos. El caso de FOXP2 resulta, una vez más, especialmente ilustrativo. El gen está sujeto a una maduración alternativa en todas las especies examinadas hasta el momento74,75, de modo que da lugar a distintas isoformas proteínicas que parecen desempeñar diferentes papeles fisiológicos76. Por otra parte, y tal como se apuntó en el apartado anterior, la proteína FOXP2 parece actuar integrada en un complejo multiproteínico del que también formarían parte, cuando mínimo, FOXP1, FOXP4 y CtBP1. Finalmente, y puesto que al menos la isoforma principal funciona como un factor transcripcional, la proteína deberá translocarse al núcleo celular para poder desarrollar dicha función, la cual exige además su unión previa a otros factores proteínicos28.
- –
Adicionalmente, el grado en que un gen concreto contribuye a un determinado proceso biológico durante el desarrollo depende crucialmente del momento, el lugar y la cantidad en que se sintetiza el producto (o productos) que codifica. Diversos mecanismos moleculares, de extremada complejidad (secuencias reguladoras en cis, factores transcripcionales y traduccionales de carácter proteínico que actúan en trans, ARN no codificadores [ARNnc] que funcionan como reguladores per se o confiriendo especificidad a los de naturaleza proteínica) modulan con gran precisión (y flexibilidad) tales parámetros, y de ahí también el carácter pleiotrópico de la mayoría de los genes, al que se aludió en el apartado anterior.
- –
Por otro lado, en la mayor parte de los casos (y especialmente en el de genes reguladores como FOXP2 [véase arriba]) los productos génicos no actúan de forma aislada. Más allá de su integración, ya discutida, en determinados complejos proteínico, dichos productos suelen formar parte de complejas redes reguladoras77, de ahí también el carácter poligénico de la mayor parte de los rasgos fenotípicos (recordemos que son muchos los genes cuya mutación afecta al lenguaje, pero muchos también, los genes candidato o de riesgo para cada trastorno concreto). Dentro de este tipo de redes la importancia del papel particular desempeñado por cada uno de los elementos implicados se ve claramente sobrepasada por el efecto conjunto debido al preciso equilibrio que mantienen, en un momento y lugar concretos, la totalidad de los productos codificados por el conjunto de genes implicados, los cuales se disponen generalmente en forma de gradientes o de combinaciones específicas de moléculas señalizadoras, que poseen propiedades fisicoquímicas de especial interés en relación con la regulación de los procesos de desarrollo (véase más adelante). El efecto homeostático ejercido por los restantes componentes de la red y/o la presencia de modificaciones adicionales en otros puntos de la misma vuelve difícilmente predecible (y significativamente variable) el fenotipo asociado a la mutación de un gen particular (recordemos que la mutación de algunas de las dianas de FOXP2 daba lugar a trastornos cognitivos de naturaleza diferente al asociado a la mutación del propio gen). Consecuentemente, ni siquiera la presencia de una misma proteína en un mismo lugar y en un mismo momento del desarrollo puede ser suficiente para garantizar que el fenotipo a cuya aparición contribuye en un determinado individuo o población sea el mismo en otros individuos o poblaciones diferentes.
- –
Asimismo, y dado que la célula en la que se expresa un gen no constituye un sistema aislado, dicha expresión se verá sometida al efecto constante de multitud de factores externos, que conciernen, por un lado, a los restantes niveles de complejidad que cabe distinguir en el sustrato neuronal del lenguaje desde el punto de vista neurobiológico (tejidos, circuitos, áreas cerebrales, etc.), pero que en último término proceden del ambiente en que transcurre el desarrollo. Dichos factores, endógenos y exógenos, no sólo modifican el patrón de expresión de los genes a través de rutas transductoras de información que activan cascadas reguladoras integradas por factores transcripcionales y traduccionales, sino que pueden originar en los propios genes modificaciones epigenéticas (esto es, alteraciones heredables de la estructura del ADN debidas a la metilación de ciertas bases o a la modificación de las proteínas responsables de su organización) que afectan también a su expresión. Cada vez más, los factores epigenéticos se consideran un mecanismo generalizado y estratégico de regulación de las funciones cognitivas y el comportamiento78. Cada vez más también, parece que lo especialmente relevante en lo concerniente al desarrollo y a la emergencia de cualquier rasgo fenotípico es el estado transcripcional de la célula (esto es, qué ARN y en qué cantidades se hallan presentes, y qué funciones desempeñan)71. Al margen de la epigénesis, existen otros factores adicionales, también heredables, que regulan el desarrollo de los organismos, especialmente durante las primeras etapas, entre los que cabe destacar los gradientes proteínicos heredados por vía materna79.
- –
Adicionalmente, la existencia de variación somática puede conducir a la aparición de poblaciones celulares genotípicamente diferentes que expresen distintas variantes de un mismo gen (en el escenario más simple, una versión mutada y la silvestre). La incidencia de este fenómeno, asociado a la actividad de ciertos tipos de transposones, podría ser especialmente relevante durante (y para) el desarrollo del sistema nervioso, condicionando fenómenos tan significativos como la neurogénesis y la función neuronal, y en último término, la plasticidad de las neuronas80.
- –
Por lo demás, diversos factores generales, de carácter fisicoquímico, modulan también de una manera decisiva los procesos de desarrollo, condicionando el itinerario ontogenético de cualquier rasgo fenotípico (entre otros cabe mencionar la viscoelasticidad, la difusión y la oscilación bioquímicas diferenciales, la dinámica de los gradientes de sedimentación y difusión, la excitabilidad mecanoquímica o las dimensiones del propio espacio en que transcurren las reacciones químicas, que actúan en combinación con propiedades básicas de las células, como la polaridad y la adhesión diferencial81,82). Tales factores determinan crucialmente la actuación de los restantes elementos reguladores implicados (proteínas, ARN, hormonas, etc.), llegando a explicar dimensiones básicas de la organización de los tejidos en desarrollo, como la regionalización o la aparición de regularidades morfológicas72,83.
- –
Finalmente, en todo proceso de desarrollo existe un alto componente estocástico, que remite en último término a la interacción azarosa entre las moléculas implicadas. De ahí, por un lado, que procesos de desarrollo idénticos que transcurran en ambientes equivalentes puedan dar lugar a fenotipos diferentes84, mientras que, por otro, el azar desempeñe también un papel crucial en la propia evolución de las relaciones genotipo-fenotipo72. La importancia de este tipo de efectos estocásticos se ha demostrado especialmente significativa para el desarrollo del cerebro84.
A la luz de todo lo discutido anteriormente, parece que lo que se ha venido en llamar «la caja negra» del desarrollo85 sería lo que explicaría, en último término, que un mismo genotipo pueda dar lugar a fenotipos diferentes (plasticidad fenotípica), pero también que un mismo fenotipo pueda ser el resultado de genotipos distintos (canalización) (fig. 1). Consecuentemente, todo lo anterior debería también permitirnos interpretar el desconcertante escenario resultante del análisis genético de los trastornos (específicos) del lenguaje, contribuyendo de este modo a establecer su genuina etiología.
Así, en un contexto necesariamente pleiotrópico, la mutación de un determinado gen puede afectar al normal desarrollo de dos (o más) estructuras cerebrales diferentes, originando anomalías estructurales y/o funcionales en dos (o más) dispositivos cerebrales distintos, lo que se traducirá en la aparición de dos (o más) déficits de procesamiento diferentes. A su vez, dichos déficits se manifestarán en forma de síntomas diversos a nivel fenotípico, que serán susceptibles de categorizarse clínicamente como correspondientes a dos (o más) trastornos distintos, que en ocasiones serán heterogéneos y en ocasiones, comórbidos. Ahora bien, como el contexto es simultáneamente poligénico, puede suceder que en determinados casos la mutación de dos (o más) genes funcionalmente relacionados afecte al desarrollo de una misma estructura cerebral, de modo que se originen anomalías estructurales y funcionales semejantes en un mismo dispositivo neuronal y por consiguiente, un único déficit de procesamiento. En este caso, lo que se advertirá a nivel fenotípico será la presencia de síntomas parecidos, susceptibles de categorizarse clínicamente como correspondientes a un único trastorno, que en ocasiones podrá ser heterogéneo. Sin embargo, puede ocurrir también (como sucede precisamente con FOXP2 y sus dianas) que la mutación de dos (o más) de tales genes funcionalmente relacionados altere el normal desarrollo de dos (o más) estructuras cerebrales diferentes, con lo que las anomalías estructurales y funcionales afectarán en este caso a dos (o más) dispositivos neuronales distintos, dando lugar a dos (o más) déficits de procesamiento diferentes. La consecuencia será que a nivel fenotípico se advertirán síntomas diversos, susceptibles de categorizarse clínicamente como correspondientes a dos (o más) trastornos distintos, que en determinados casos serán heterogéneos y en otros, comórbidos. Finalmente, conviene recordar que la contribución de cada producto disfuncional o afuncional al fenotipo anómalo está siempre condicionada por el efecto sutil ejercido por los restantes genes implicados, así como por los restantes factores moduladores del desarrollo también implicados (epigenéticos, maternos, ontogenéticos, generales, ambientales, etc.). En consecuencia, puede suceder que la mutación de un mismo gen dé lugar, en diferentes individuos o poblaciones, a diversos grados de afectación en lo concerniente a la integridad estructural y funcional de determinadas áreas cerebrales, y por consiguiente, a déficits de procesamiento y perfiles cognitivos variables, y en particular, a síntomas diversos, los cuales serán susceptibles de categorizarse clínicamente como dos (o más) subtipos del mismo trastorno o incluso como dos (o más) trastornos diferentes, que en ocasiones serán comórbidos. Pero por razones semejantes, puede ocurrir igualmente que la mutación de dos (o más) genes diferentes origine, en determinados individuos o poblaciones, anomalías estructurales y funcionales semejantes en una misma (o en diversas) área(s) cerebral(es), y por consiguiente, a déficits de procesamiento semejantes y a un perfil cognitivo similar, y en último término, a síntomas parecidos, susceptibles de categorizarse clínicamente como el mismo trastorno o como subtipos diferentes de un mismo trastorno.
FinanciaciónEste trabajo ha sido realizado al amparo del proyecto de investigación «Biolinguística: evolución, desarrollo y fósiles del lenguaje» (FFI2010-14955/FILO), subvencionado por el Ministerio de Educación y Ciencia, con financiación parcial FEDER.
Conflictos de interesesLos autores declaran no tener ningún conflicto de intereses.

