



La enfermedad de Fabry (EF) es un trastorno de depósito lisosomal, ligado al cromosoma X, resultante de la deficiencia o ausencia de la enzima alfa galactosidasa A (α-galA). El compromiso orgánico en los hombres es bien conocido, pero en las mujeres es motivo de controversia, debido en parte al fenómeno de lionización. El objetivo de este estudio fue describir el compromiso renal en una población de 35 mujeres en el momento del diagnóstico de la EF.
MétodoSe evaluaron 35 mujeres de 3 centros de referencia de Argentina. La actividad de la enzima α-galA se realizó en papel de filtro por método fluorométrico y el estudio mutacional mediante MLPA y secuenciación. Se calculó la tasa de filtrado glomerular por fórmula CKD-EPI en pacientes adultos y Schwartz en pediátricos. Se consideró albuminuria y proteinuria en al menos 2 muestras diferentes de orina en todos los casos, y a las categorías del filtrado glomerular según guías KDIGO 2012.
ResultadosEl promedio de edad del grupo completo (n=35) fue de 26,6±16,9 años; de las cuales 22 mujeres eran mayores de 18 años y 13 eran menores. El dosaje enzimático de α-galA se realizó en 29/35 pacientes y fue normal en 24/29 (82,8%). Se hallaron 7 mutaciones del gen GLA. Los resultados mostraron hiperfiltración glomerular (42,9%), pérdida urinaria de proteínas (45,7%) y disminución del filtrado glomerular (31,4%).
ConclusionesEl compromiso renal en mujeres con EF puede ser tan severo como en los hombres. El análisis de este grupo de pacientes demostró una proporción significativa de mujeres con afectación temprana evidenciada por hiperfiltración renal, albuminuria, proteinuria y caída del filtrado glomerular al momento del diagnóstico de la EF.
Fabry disease (FD) is an X-linked lysosomal storage disorder resulting from the deficiency or absence of the alpha galactosidase A (α-galA) enzyme. Organ involvement in men is well known, but in women it is controversial, partly due to random X-chromosome inactivation (Lyon hypothesis). The aim of this study was to describe renal involvement in a population of 35 women at the time of FD diagnosis.
MethodThirty-five females were evaluated in three reference centres in Argentina. The activity of the α-galA enzyme was determined on filter paper by a fluorometric method, and the mutational study by MLPA and sequencing. Glomerular filtration rate was calculated usinjg the CKD-EPI formula in adult patients and Schwartz formula in paediatric patients. Albuminuria and proteinuria were observed in at least two different urine samples in all cases, as well as the glomerular filtration rates categories according to KDIGO 2012 guidelines.
ResultsMean age of the complete group (n=35) was 26.6±16.9 years, of whom 22 were adult women (over 18) and 13 were paediatric patients. Enzymatic activity of α-galA was performed in 29/35 patients, which was normal in 24/29 (82.8%). Seven different mutations of the GLA gene were found. The results showed glomerular hyperfiltration (42.9%), urinary protein loss (45.7%), and decreased glomerular filtration rate (31.4%).
ConclusionsRenal involvement in females with FD may be as severe as in men. The analysis of this group of patients showed a significant proportion of females with early kidney damage demonstrated by renal hyperfiltration, albuminuria, proteinuria and glomerular filtration rate decreased, at the time of diagnosis of FD.
La enfermedad de Fabry (EF) es provocada por la acumulación lisosomal de glicoesfingolípidos complejos, principalmente globotriaosilceramida (GL-3) y sus metabolitos1. Dicho depósito dispara cascadas fisiopatogénicas en el endotelio vascular y células de diferentes tejidos (cardíacas, renales y nerviosas, entre otras) que conducen a la muerte celular, con progresión a fibrosis y deterioro orgánico irreversible2,3. El almacenamiento de globotriaosilceramida se debe a la actividad deficiente o nula de la α-galactosidasa A (α-galA, EC 3.2.1.22). El gen GLA, que codifica la α-galA, está ubicado en el cromosoma X (Xq22.1), por lo cual prácticamente todos los hombres portadores de una mutación genética (hemicigotos) desarrollan la enfermedad, mientras que las mujeres (heterocigotas) presentan una amplia variabilidad en la severidad de su fenotipo, debido principalmente a la inactivación aleatoria de uno de los cromosomas X en cada una de sus células (hipótesis de Lyon)4. La intensidad de los síntomas va a depender, en mayor parte, de la actividad residual de la enzima α-galA. El dosaje de la misma se realiza en gota de sangre seca en papel de filtro; en varones la actividad disminuida confirma la enfermedad, mientras que en mujeres se debe recurrir al estudio molecular, por la existencia de falsos negativos.
Las manifestaciones de la EF son multisistémicas y comienzan en la infancia, alcanzando una afectación severa en la tercera o cuarta décadas de la vida. Las principales manifestaciones son las características acroparestesias en las manos y los pies, síntomas gastrointestinales, angioqueratomas, dishidrosis, intolerancia al ejercicio y al calor, pérdida de la audición, arritmias, miocardiopatía hipertrófica, accidentes cerebrovasculares e insuficiencia renal5,6.
Si bien es una enfermedad conocida desde hace más de 100 años, durante la última década el pronóstico de la EF ha dado un giro significativo debido a la posibilidad de terapia de reemplazo enzimático7,8.
En 2001 se describió que la mayoría de las mujeres eran asintomáticas, con una calidad de vida completamente normal o desarrollaban solo manifestaciones leves de la enfermedad9. Sin embargo, estudios posteriores han reportado que las mujeres heterocigotas pueden desarrollar síntomas severos de la EF y tener riesgo de muerte prematura10,11.
La EF es panétnica y, dada su baja incidencia, no se ha podido describir con exactitud su prevalencia, que varía entre 1:40.000 hombres a 1:117.000 nacidos vivos9,12. La gran variabilidad fenotípica, junto con la sintomatología inespecífica, dificultan significativamente el diagnóstico. Al igual que otras enfermedades raras ha sido difícil comprender la progresión natural de la EF debido a la escasa información clínica, sobre todo en mujeres.
El objetivo de este estudio fue describir el compromiso renal de la EF en mujeres al momento del diagnóstico.
Material y métodosEstudio descriptivo, transversal y multicéntrico. Se evaluaron 35 mujeres con EF de 3 centros de referencia en Argentina: Servicio de Terapia Intensiva del Hospital Dr. Enrique Erill de Escobar, Centro de Neurociencias Los Manantiales del Grupo Gamma de Rosario y Centro de Infusión y Estudio de Enfermedades Lisosomales del Instituto de Nefrología Pergamino. La actividad de la enzima α-galA se realizó en papel de filtro por método fluorométrico en Laboratorio de Neuroquímica Dr. N. A. Chamoles/FESEN (Buenos Aires, Argentina)13 y el estudio mutacional mediante MLPA y secuenciación en Medical Genetics Laboratories-Baylor College of Medicine (Houston, TX, EE. UU.)14,15. No se incluyeron en el estudio las pacientes con riesgo de enfermedad glomerular por otra causa diferente a la EF.
El compromiso renal de la población en estudio fue determinado por la pérdida de proteínas en orina (albuminuria/proteinuria), la presencia de hiperfiltración glomerular y/o la disminución de la tasa de filtrado glomerular estimada (TFGe). No se evaluó función tubular por no disponer de datos.
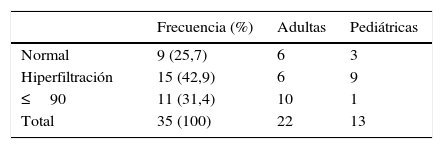
Se calculó la TFGe utilizando la creatinina sérica, por fórmula CKD-EPI en los pacientes adultos y Schwartz en pediátricos16,17. Ambas fórmulas de TFGe han sido recientemente validadas en pacientes con EF18,19. Valores superiores a 125ml/min/1,73m2 se definieron como hiperfiltración glomerular18. Las categorías del filtrado glomerular fueron determinadas según las guías internacionales del consorcio KDIGO 2012, como se observa en la tabla 120.
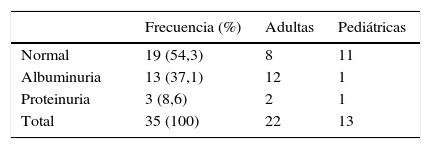
Se evaluó albuminuria y proteinuria de toda la población del estudio. La albúmina en orina fue analizada por inmunoturbidimetría21 y la proteinuria mediante método colorimétrico22. Se consideró albuminuria patológica a valores>30mg/día o>30mg/g de creatinina, y proteinuria a valores>300mg/día o>300mg/g de creatinina, en al menos 2 muestras diferentes de orina en todos los casos.
El trabajo de investigación fue aprobado por cada comité de ética local, y se tomó consentimiento informado por escrito de cada paciente del estudio.
Análisis estadísticoLas variables categóricas se expresaron como números (porcentajes) y las variables continuas como media±desviación estándar (DE). Las tablas de contingencia se analizaron con la prueba exacta de Fisher. Las diferencias se consideraron significativas si p<0,05. Los análisis estadísticos se realizaron con el software GraphPad Prism 2.0 (GraphPad, San Diego, EE. UU.).
ResultadosDatos demográficosSe estudiaron 35 mujeres con EF con un promedio de edad de 26,6±16,6 años. Del total de la población estudiada 22 mujeres eran adultas (mayores a 18 años) y 13 eran menores, con un promedio de 37,0±12,2 y 9,2±4,3 años respectivamente. Se detectaron 2 casos índices; ambas mujeres adultas de 28 y 55 años.
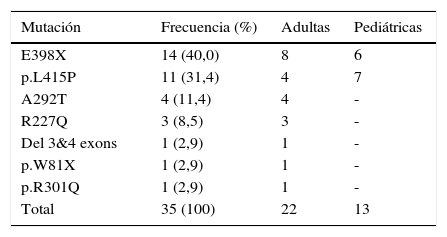
MutacionesSe detectaron 7 mutaciones del gen GLA (tabla 2). La más frecuente fue la mutación E398X, con un total de 14 pacientes (40,0%); 6 de edades pediátricas y 8 adultas. Le sigue en frecuencia la mutación p.L415P con 11 pacientes (31,4%); 7 pacientes pediátricas y 4 adultas. En menor porcentaje encontramos las mutaciones A292T (11,4%), R227Q (8,5%), deleción de exones 3&4 (2,9%), p.W81X (2,9%) y p.R301Q (2,9%). Todas mutaciones clásicas, a excepción de la mutación p.R301Q (n=1), que está descrita como causante de fenotipo clásico y también como variante tardía de la EF (http://fabry-database.org/mutants). El total de las pacientes de cada genotipo pertenece a la misma familia.
Dosaje enzimáticoSe ha realizado dosaje enzimático de α-galA a 29/35 pacientes (82,9%) y fue normal en 24/29 (82,8%). No se dispone de los resultados de 6 pacientes. Los dosajes disminuidos correspondieron a las mutaciones E398X (n=2) y p.L415P (n=3).
Compromiso renalEn las tablas 3 y 4 se describen la TFGe (ml/min/1,73 m2) y la pérdida urinaria de proteínas (albuminuria/proteinuria) de la población del estudio.
El 25,7% (n=9) de las pacientes tenía función renal normal en el momento del diagnóstico de la EF. El 42,9% (n=15) presentó hiperfiltración glomerular. Disminución de la TFGe (<90ml/min/1,73m2) fue hallada en 11 pacientes (31.4%), 10 adultas y una pediátrica. De las pacientes adultas con compromiso renal 9 eran de categoría G2 y una de categoría G3a, mientras que la paciente pediátrica se encontraba en categoría G2. La disminución de la TGFe se presentó predominantemente en pacientes adultas (test de Fisher, p=0,0284).
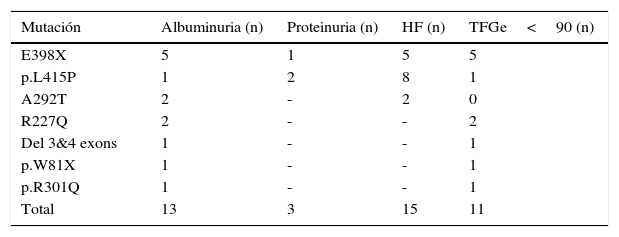
Se objetivó pérdida urinaria patológica de proteínas en 16/35 (45,7%) mujeres, con predominio de las pacientes adultas (test de Fisher, p=0,0125). Fue detectada albuminuria en 12 pacientes adultas y en una paciente pediátrica (37,1%), y proteinuria en 2 adultas y en una niña (8,6%). En la tabla 5 se observa la relación de la pérdida urinaria de proteínas (albuminuria/proteinuria) y la TFGe con el tipo de mutación.
Relación de la pérdida urinaria de proteínas y TFGe con el genotipo
| Mutación | Albuminuria (n) | Proteinuria (n) | HF (n) | TFGe<90 (n) |
|---|---|---|---|---|
| E398X | 5 | 1 | 5 | 5 |
| p.L415P | 1 | 2 | 8 | 1 |
| A292T | 2 | - | 2 | 0 |
| R227Q | 2 | - | - | 2 |
| Del 3&4 exons | 1 | - | - | 1 |
| p.W81X | 1 | - | - | 1 |
| p.R301Q | 1 | - | - | 1 |
| Total | 13 | 3 | 15 | 11 |
HF: hiperfiltración; TFGe: tasa de filtrado glomerular estimada en ml/min/1,73m2.
De manera similar a otras enfermedades de herencia ligada al cromosoma X, en la EF las complicaciones son menos frecuentes y más variables en severidad en mujeres que en varones, aunque algunas mujeres pueden presentar fenotipos similares a estos últimos6,11,23.
La actividad enzimática en las mujeres con EF suele estar en el rango normal inferior, debido al patrón de herencia ligado al cromosoma X24. Por lo tanto, la determinación de la actividad plasmática de α-galA en las mujeres solo puede indicar la posible presencia de la EF, pero no establece ni descarta la enfermedad. Esta es la razón por la cual se recomienda el estudio genético en mujeres. La variabilidad de inactivación del X (hipótesis de Lyon) podría explicar en parte el alto porcentaje de dosajes enzimáticos normales encontrados en nuestra población. Esta inactivación cromosómica impacta significativamente en el fenotipo y en la historia natural de la EF en mujeres25.
La nefropatía es una de las principales complicaciones de la EF26. Se caracteriza por niveles variables de gravedad, con una tasa de progresión de ERC similar a la nefropatía diabética, con evidencias que sugieren que los pacientes no tratados suelen desarrollar enfermedad renal terminal hacia la cuarta década de la vida27. Por lo general, los pacientes sin tratamiento muestran 3 fases clínicas de la nefropatía Fabry. La primera ocurre en la infancia y adolescencia, y se caracteriza por hiperfiltración glomerular. La segunda fase clínica por presentar proteinuria, lipiduria, cristales en «cruz de Malta» en el sedimento de orina examinado por microscopía polarizada, alteración de la capacidad de concentración o dilución de la orina y otras disfunciones renales. En la tercera fase se presenta enfermedad renal grave y progresiva, con compromiso cardiovascular y cerebral2. Es conocido que el compromiso renal es más frecuente en hombres que en mujeres. En nuestro reporte destacamos un alto porcentaje de mujeres con hiperfiltración renal, con predominio en las pacientes en edad pediátrica, como probable compensación glomerular ante la nefropatía Fabry. Por otra parte, la caída de la TFGe se evidenció en un tercio de nuestra población, con significación estadística en las pacientes adultas sobre las pediátricas, sin terapia específica para la EF. Esto podría interpretarse como la progresión natural de la nefropatía. Una limitación de este estudio es que se valoró la TFGe por fórmula y no medida.
La excreción urinaria de proteínas está directamente asociada con la progresión de la enfermedad renal en hombres y mujeres con EF28. La albuminuria y la proteinuria son elementos comunes de la progresión de la nefropatía Fabry, que pueden alcanzar rango nefrótico29. En el presente estudio evidenciamos en casi la mitad de las pacientes pérdida urinaria de proteínas (albuminuria/proteinuria), con predominio en la población adulta, al momento del diagnóstico de la EF. La albuminuria fue el signo de nefropatía más relevante detectado. Estos resultados, al igual que otros estudios, como los publicados en 2008 por Wilcox et al.23 y en 2006 por Deegan et al.30, muestran que las mujeres con EF tienen un riesgo significativo de desarrollar compromiso renal, tan severo como el descrito en hombres.
ConclusiónLos principales hallazgos de nuestro estudio fueron: 1) los dosajes enzimáticos de α-galA fueron normales en un 82,8%; 2) el 42.9% de las pacientes presentó hiperfiltración glomerular; 3) la disminución de la TFGe se evidenció predominantemente en mujeres adultas; 4) se objetivó pérdida de proteínas en orina en un 45,7% de las pacientes, con predominio en la población adulta; y 5) la albuminuria fue el signo de nefropatía más relevante dentro de las pérdida urinaria de proteínas.
El compromiso renal en mujeres con EF puede ser tan severo como en los hombres23. Este análisis demostró una proporción significativa de mujeres con afectación temprana evidenciada por hiperfiltración renal, pérdida urinaria de proteínas (albuminuria/proteinuria) y caída del filtrado glomerular al momento del diagnóstico de la EF. Se destaca la importancia de un diagnóstico temprano y un seguimiento periódico y exhaustivo, con el fin de evitar el daño orgánico irreversible.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran que no tienen conflicto de intereses potenciales relacionados con los contenidos de este artículo.