

Las enfermedades por depósito lisosomal son un grupo heterogéneo de unos 50 trastornos metabólicos hereditarios.
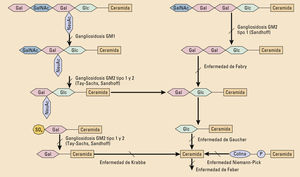
EtiopatogeniaEstos trastornos tienen en común un déficit genético de enzimas u otras proteínas relacionadas con la hidrólisis de macromoléculas en los lisosomas. Salvo tres, que se heredan ligadas al cromosoma X, las demás tienen un patrón de herencia autosómico recesivo. Se clasifican por el tipo de sustrato que se acumula en los lisosomas.
EpidemiologíaPor su prevalencia, se las considera enfermedades raras, aunque agrupadas pueden afectar hasta 1/5.000 nacidos vivos. Entre ellas, destacan por su frecuencia la enfermedad de Gaucher y la enfermedad de Fabry.
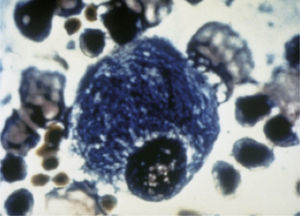
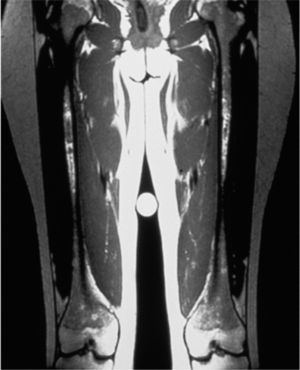
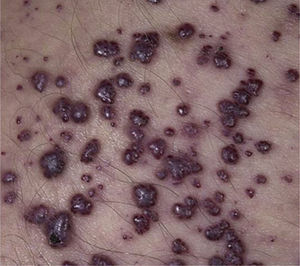
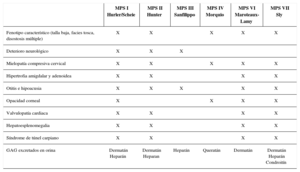
Manifestaciones clínicasSu sintomatología es multisistémica y, salvo por la existencia en algunas de signos típicos dismorfológicos, en general, son difíciles de diagnosticar.
PronósticoLa mayoría presenta un curso natural hacia la discapacidad y la muerte.
TratamientoA la espera de la terapia génica, muchas de ellas son ahora susceptibles de tratamientos específicos con sustitución enzimática.
Palabras clave
Lysosomal storage disorders embrace a heterogeneous group of more than 50 different genetic diseases.
PathogenesisLysosomal storage disorders could be explained by specific enzyme or protein deficiencies related with lysosomal hydrolysis. Most of them are inherited as autosomal recessive traits, but tree conditions have X-linked inheritance. Classification is based on the kind of substrate that lysosomes accumulate.
EpidemiologyThe prevalence values for individual lysosomal storage disorders clearly define these as rare genetic disorders. However, when taken as a group, lysosomal storage disorders are more common, with a prevalence of 1/5,000 live births. Gaucher and Fabry diseases are the most frequent.
Clinical manifestationsDiagnosis is difficult because disease presentation may be subtle, except for some typical dysmorphological features.
PrognosisPrognosis is severe with a natural course to disability and death in most patients
TreatmentSpecific enzyme replacement therapy is available for many lysosomal storage disorders. Meanwhile, gene therapeutic techniques are being developed.
Keywords
Identifíquese
¿Aún no es suscriptor de la revista?

Comprar el acceso al artículo
Comprando el artículo el pdf del mismo podrá ser descargado
Precio: 19,34 €
Teléfono para incidencias
De lunes a viernes de 9h a 18h (GMT+1) excepto los meses de julio y agosto que será de 9 a 15h