




La enfermedad de Creutzfeldt-Jakob (ECJ) es la encefalopatía espongiforme transmisible más frecuente en humanos. Se caracteriza clínicamente por una demencia rápidamente progresiva, mioclonías y ataxia. Las formas familiares son poco frecuentes (5-15% de los casos) y se presentan en ocasiones con síntomas atípicos y con un curso clínico más prolongado que la forma esporádica. Las pruebas complementarias recomendadas ante la sospecha clínica de ECJ son la resonancia magnética craneal, el electroencefalograma, la determinación de la proteína 14-3-3 en líquido cefalorraquídeo (LCR) y el estudio del gen de la proteína priónica (PRNP).
Se expone el caso de una paciente de 54 años que presentó un trastorno depresivo prolongado asociado a un deterioro neurológico progresivo. Entre las pruebas complementarias, la elevación de la proteína 14-3-3 en LCR y el hallazgo de la mutación E200K en el estudio genético fueron esenciales para llegar al diagnóstico de una ECJ familiar.
Creutzfeldt-Jakob disease (CJD) is the most frequent of the human transmissible spongiform encephalopathies. It is clinically characterized by rapidly progressive dementia, myoclonia and ataxia. Familial CJD is uncommon (5-15% of cases) and sometimes it has atypical symptoms and longer clinical course than sporadic CJD. Diagnostic tests recommended when patients present with a clinical picture of CJD are: brain magnetic resonance imaging, electroencephalogram, 14-3-3 protein determination in the cerebrospinal fluid and study of the prionic protein gene (PRNP).
The case of a 54-year-old female patient with prolonged depression added to a progressive neurological impairment is presented. Among the complementary tests, cerebrospinal fluid 14-3-3 protein high level and E200K mutation discovered in genetic study were essential to support the diagnosis of familial CJD.
La enfermedad de Creutzfeldt-Jakob (ECJ) es una encefalopatía espongiforme producida por priones que presenta una incidencia de 0,5 a 2 casos por millón de habitantes y año1. Se han descrito 4 grupos de encefalopatía espongiforme transmisible en humanos: kuru, enfermedad de Creutzfeldt-Jakob, síndrome de Gerstmann-Sträussler-Scheinke e insomnio familiar fatal. La ECJ es la más frecuente y presenta 4 tipos, según su modo de adquisición: esporádica, iatrógena o adquirida, familiar y la nueva variante, asociada al consumo de alimentos procedentes de animales infectados. La ECJ esporádica es la más prevalente (80-90% de los casos) y se manifiesta como una encefalopatía mioclónica subaguda (demencia rápidamente progresiva, mioclonías y ataxia) con una duración media de 5 a 12 meses2-5. La ECJ familiar supone sólo el 5-15% de los casos y, en general, sus síntomas son similares a los de la forma esporádica, aunque se han descrito manifestaciones clínicas poco habituales y su duración media es superior, de 1 a 5 años6. A continuación se expone el caso clínico de una paciente que presentó un trastorno depresivo resistente al tratamiento seguido de un deterioro neurológico progresivo que orientó el diagnóstico de ECJ familiar.
Exposición del casoMujer de 54 años sin alergias conocidas que presenta como antecedentes personales gastritis superficial asociada a infección por Helicobacter pylori erradicada, osteoporosis, hipoparatiroidismo y cefalea crónica diaria asociada a abuso de analgésicos. No es fumadora ni bebedora habitual. La paciente está en tratamiento con ácido alendrónico semanal, carbonato cálcico asociado a colecalciferol y omeprazol diarios y analgésicos (paracetamol, metamizol) y almotriptán, que toma a demanda.
En junio de 2008 acude a la consulta de atención primaria por presentar un cambio en el estado de ánimo con anhedonia, apatía, inestabilidad emocional e insomnio, que relaciona con problemas personales. Se inicia tratamiento con escitalopram 20mg y lorazepam 1mg sin observarse mejoría clínica, por lo que se asocia amitriptilina 25mg. Tampoco se obtiene respuesta a este tratamiento, por lo que se realizan los siguientes exámenes complementarios, que resultaron negativos: hemograma, velocidad de sedimentación globular, bioquímica de sangre, sedimento de orina, serología de sífilis y virus de inmunodeficiencia humana, hormonas tiroideas, vitamina B12, ácido fólico, electroencefalograma y tomografía axial computarizada (TAC) cerebral. En un periodo de 12 meses la paciente recibe asistencia del servicio de psiquiatría, que pauta diversos tratamientos antidepresivos y ansiolíticos, sin que se observe mejoría anímica. Al cuadro inicial se añaden otros síntomas como anorexia, pérdida de peso (9kg en un año), dolores inespecíficos en miembros superiores e inferiores, hipoestesias en miembros inferiores, mioclonías sensibles a estímulos auditivos y táctiles y marcha inestable progresiva. Como consecuencia de la inestabilidad, la paciente presenta una caída accidental con traumatismo craneoencefálico asociado. En la TAC cerebral que se realiza durante el ingreso se observa una lesión de 6mm isquémica antigua en ganglios basales izquierdos. Al alta con el diagnóstico de traumatismo craneoencefálico y accidente cerebrovascular antiguo se pauta ácido acetilsalicílico 100mg diario.
Otras pruebas complementarias que resultan normales son el eco-Doppler de miembros inferiores y el electromiograma de miembros superiores e inferiores. El estudio de potenciales evocados somatosensoriales (PESS) muestra un trastorno bilateral de la conducción somatosensorial en miembros superiores e inferiores.
La resonancia magnética (RM) craneal detecta pequeñas alteraciones focales en sustancia blanca cortico-subcortical frontal y parietal izquierda y alguna alteración puntiforme frontal derecha de probable origen vascular.
La RM cervical muestra 2 hernias discales entre C4-C5 y C5-C6 con compresión medular y signos de mielopatía cervical por compresión a dicho nivel. A partir de este hallazgo, se decide abordaje quirúrgico con doble discectomía cervical y artrodesis intersomática. Tras la cirugía no se observa mejoría del trastorno sensitivo ni motor con la rehabilitación y la paciente presenta deterioro progresivo de sus funciones superiores, imposibilidad para la deambulación y bipedestación, demencia con afasia, apraxia y agnosia e incontinencia urinaria. Ante la sospecha de ECJ se realiza un electroencefalograma (EEG) y una punción lumbar. El EEG muestra un patrón de enlentecimiento difuso inespecífico. El análisis bioquímico, citológico y microbiológico del LCR es normal, pero se demuestra la presencia de la proteína 14-3-3. En el estudio genético de la proteína priónica se evidencia la mutación E200K y un polimorfismo heterocigoto metionina/valina en el codón 129 (129M/V). Estos resultados sugieren que se trata de una encefalopatía espongiforme transmisible de origen genético, por lo que se recomienda estudio genético de familiares.
DiscusiónLa ECJ familiar es una enfermedad hereditaria con patrón de herencia autosómico dominante que representa un 5-15% del total de casos de ECJ. En estas familias se han descrito numerosas mutaciones en el gen de la proteína priónica, denominado PRNP y situado en el cromosoma 20. Respecto a la penetrancia de la mutación, existe discrepancia en los resultados de distintos estudios, de manera que algunos apoyan la penetrancia completa y otros defienden una penetrancia incompleta1.
Desde el punto de vista clínico, la ECJ esporádica cursa como una encefalopatía mioclónica subaguda con una duración media de 5 a 12 meses, siendo excepcional la duración superior a los 2 años. En la mayoría de los casos se distinguen 3 etapas en el curso clínico1:
- •
Un periodo inicial, caracterizado por síntomas psíquicos como apatía, tristeza y déficit de atención.
- •
Un periodo de estado, en el que se produce un rápido deterioro global con una progresiva pérdida de relación con el entorno y una dependencia absoluta.
- •
Un periodo final, en el que se desarrolla un mutismo acinético y desconexión del medio hasta el fallecimiento, generalmente como consecuencia de sepsis por infecciones recurrentes.
Las mioclonías son un signo característico en estos pacientes. Suelen ser multifocales, asincrónicas y arrítmicas, y aparecen de forma espontánea o tras estímulos táctiles o auditivos.
La ECJ familiar suele manifestarse con unos síntomas y signos similares, aunque tiene mayor diversidad clínica, suele iniciarse a edades inferiores y presenta una duración media mayor, de 1 a 5 años1-5. En nuestro caso, la paciente presentó inicialmente un trastorno del estado de ánimo que fue seguido por un deterioro neurológico progresivo con mioclonías, alteraciones sensitivas e inestabilidad de la marcha, con una duración aproximada de 15 meses hasta alcanzar el periodo final de mutismo acinético y desconexión del medio.
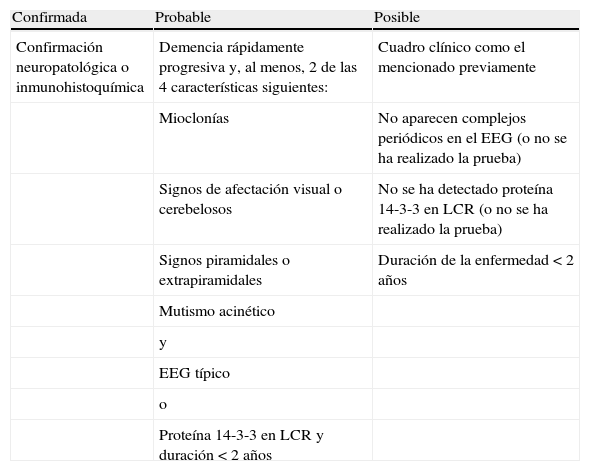
El diagnóstico de la ECJ se basa en la presentación clínica, su curso progresivo y la ausencia de un diagnóstico alternativo. Aunque la OMS ha establecido unos criterios diagnósticos de ECJ esporádica posible, probable o confirmada (tabla 1), no existen pruebas diagnósticas definitivas en vivo1. Sin embargo, la RM craneal, el EEG y el estudio del LCR puede hacer más seguro su diagnóstico1. Estas pruebas no son accesibles para el médico de familia, por lo que la sospecha diagnóstica debe plantearse en la consulta de atención primaria a partir de los datos recogidos en una anamnesis minuciosa y una exploración física general y neurológica.
Criterios diagnósticos de enfermedad de Creutzfeldt-Jakob esporádica
| Confirmada | Probable | Posible |
| Confirmación neuropatológica o inmunohistoquímica | Demencia rápidamente progresiva y, al menos, 2 de las 4 características siguientes: | Cuadro clínico como el mencionado previamente |
| Mioclonías | No aparecen complejos periódicos en el EEG (o no se ha realizado la prueba) | |
| Signos de afectación visual o cerebelosos | No se ha detectado proteína 14-3-3 en LCR (o no se ha realizado la prueba) | |
| Signos piramidales o extrapiramidales | Duración de la enfermedad<2 años | |
| Mutismo acinético | ||
| y | ||
| EEG típico | ||
| o | ||
| Proteína 14-3-3 en LCR y duración<2 años |
EEG: electroencefalograma; LCR: líquido cefalorraquídeo.
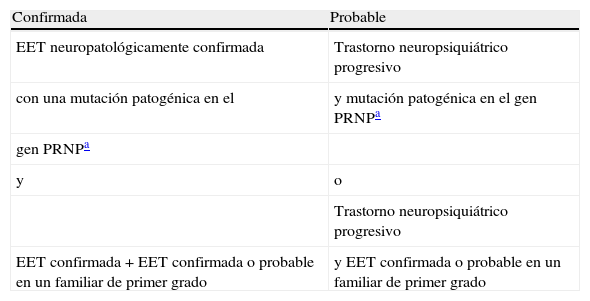
En relación con la ECJ familiar, la OMS también ha definido unos criterios diagnósticos de enfermedad probable o confirmada (tabla 2). Se establece el diagnóstico de ECJ familiar probable cuando aparece un trastorno neuropsiquiátrico progresivo y una mutación patogénica en el gen PRNP o un caso de ECJ confirmada o probable en un familiar de primer grado. Cuando se presenta una ECJ confirmada por biopsia cerebral o autopsia junto a una mutación patogénica en el gen PRNP o un caso de ECJ confirmada o probable en un familiar de primer grado se concluye el diagnóstico de ECJ familiar confirmada1.
Criterios diagnósticos de encefalopatía espongiforme transmisible genéticamente determinada
| Confirmada | Probable |
| EET neuropatológicamente confirmada | Trastorno neuropsiquiátrico progresivo |
| con una mutación patogénica en el | y mutación patogénica en el gen PRNPa |
| gen PRNPa | |
| y | o |
| Trastorno neuropsiquiátrico progresivo | |
| EET confirmada+EET confirmada o probable en un familiar de primer grado | y EET confirmada o probable en un familiar de primer grado |
ETT: encefalopatía espongiforme transmisible; PRNP: proteína priónica.
La RM craneal ha demostrado su utilidad para el diagnóstico de ECJ esporádica, donde se encuentran alteraciones en aproximadamente el 80% de los pacientes. Los hallazgos más frecuentes son la hiperseñal de los ganglios basales (cabeza del caudado y del putamen) en secuencias potenciadas en T2 o en fases de densidad protónica o fases FLAIR (fast fluid-attenuated inversion recovery). Según diversos estudios, la sensibilidad de la RM en la ECJ esporádica oscila entre el 58,3 y el 92,3%, y la especificidad entre el 81,6 y el 95%7,8. Por otro lado, en el 80% de los casos de la variante de ECJ se ha descrito una hiperintensidad bilateral en el tálamo posterior correspondiente a la región pulvinar9. Este signo del pulvinar presenta una sensibilidad del 78% y una especificidad del 100% para la variante de ECJ y ha sido incluido en la definición de caso de la OMS. En relación con la ECJ familiar, se ha estudiado menos la utilidad diagnóstica de la RM craneal, pero también se han descrito alteraciones similares a la ECJ esporádica7. En nuestra paciente se observaron pequeñas alteraciones focales de la sustancia blanca subcortical frontal y parietal izquierda y alguna puntiforme frontal derecha, que fueron inicialmente relacionadas con un posible origen vascular.
El EEG muestra típicamente complejos de ondas trifásicas que se repiten de forma periódica en dos tercios de los pacientes con ECJ esporádica1. En la forma familiar se observa este patrón en menos del 10% de los casos y está ausente en los pacientes con la variante de ECJ3. En nuestra paciente el EEG mostró un patrón de enlentecimiento difuso inespecífico que no contribuyó a concretar el diagnóstico.
El estudio del LCR es especialmente útil en el diagnóstico de la ECJ. La proteína 14-3-3 de origen neuronal está presente en muy pequeña cantidad en condiciones normales en el LCR, pero aumenta mucho su concentración en las enfermedades por priones y en otras enfermedades con destrucción neuronal muy rápida. La determinación de proteína 14-3-3 en LCR es una prueba muy sensible (94%) y específica (93%) para el diagnóstico de ECJ esporádica, aunque la sensibilidad disminuye en los casos de ECJ familiar, iatrogénica y la nueva variante. Otras enfermedades en las que puede detectarse la proteína 14-3-3 en LCR son encefalitis de Hashimoto, encefalitis por virus herpes simple y otros virus, hemorragia subaracnoidea, hipoxia cerebral, encefalopatía metabólica tras intoxicación por barbitúricos, glioblastoma, encefalopatía paraneoplásica o accidente cerebrovascular agudo. Sin embargo, teniendo en cuenta el contexto clínico de sospecha diagnóstica de ECJ, el diagnóstico diferencial con estas entidades resulta más fácil1,10.
Otra prueba de especial relevancia para el diagnóstico de la ECJ familiar es el estudio genético. Se han descrito más de 20 mutaciones que se corresponden con diversos cuadros clínicos, siendo la más frecuente en el codón 178, seguida del codón 200. La expresión de una mutación patogénica en el gen PRNP en un paciente con una enfermedad neurodegenerativa permite confirmar el diagnóstico de la enfermedad6. Además, este hallazgo está incluido entre los criterios diagnósticos propuestos por la OMS para la ECJ familiar. El polimorfismo en el codón 129 (Metionina/Valina) es el más relevante de los distintos polimorfismos del PRNP y se relaciona con distinto grado de susceptibilidad de presentar una enfermedad por priones. Mientras que en los casos de ECJ esporádica predomina el genotipo homocigoto (129M/M y 129V/V), en los otros tipos de ECJ es más habitual el genotipo heterocigoto (129M/V)1. Esto se corresponde con lo observado en el estudio genético realizado a nuestra paciente, donde se encontró la mutación E200K del gen PRNP, con un polimorfismo heterocigoto Metionina/Valina en el codón 129.
Actualmente no se conocen tratamientos eficaces para la ECJ, sin embargo, se encuentran en marcha algunos ensayos clínicos con fármacos como la quinacrina y clorpromacina, que han sido empleados en algunos pacientes como uso compasivo1.
Aunque la ECJ es una enfermedad muy infrecuente en la consulta de atención primaria, debemos sospecharla ante pacientes con cuadros psiquiátricos asociados a deterioro neurológico progresivo inesperado, con el objetivo de proporcionar una atención médica precoz y adecuada, especialmente en los cuidados en el domicilio en la fase de máxima dependencia, así como una asistencia familiar completa, incluido el consejo genético a los descendientes sanos.
