







Los tumores neuroendocrinos pueden derivar de la cresta neural (ganglioneuroma, neuroblastoma, paraganglioma), de glándulas endocrinas (adenoma de hipófisis, feocromocitoma), de islotes (medular de tiroides, tumor de Merkel cutáneo, páncreas) o de células del sistema endocrino difuso1. Éste se encuentra disperso, por orden de frecuencia, en el aparato digestivo, aparato respiratorio, timo, aparato genitourinario, área otorrinolaríngea y el resto de órganos internos, y da lugar a los tumores carcinoides (TCa) cuando éstos son bien diferenciados2.
Los tumores de los islotes pancreáticos (TNP) y los del sistema endocrino difuso gastrointestinales, definidos en la bibliografía como tumores endocrinos gastroenteropancreáticos (TEGEP), forman un grupo de tumores que presentan una serie de características comunes3. Poseen una anatomía patológica similar, suelen ser bien diferenciados y pueden secretar determinadas aminas biógenas y péptidos vasoactivos al torrente sanguíneo y causar síntomas específicos, en cuyo caso se consideran funcionantes. Su manejo terapéutico es complejo y multidisciplinario y está basado en grados de evidencia bajos4. Más del 80% de los TEGEP expresan receptores de somatostatina, lo que puede utilizarse con fines diagnósticos gammagrafía con octreótido o terapéuticos análogos de somatostatina utilizados en el síndrome funcional. Son tumores muy vascularizados, lo que también tiene implicaciones terapéuticas y en el diagnóstico por la imagen. Pueden aparecer agregados en síndromes familiares, entre los que destaca el síndrome de las neoplasias endocrinas múltiples tipo 1, de herencia autosómica dominante, con potencial afectación de múltiples órganos como las glándulas paratiroides, hipófisis, páncreas y, más raramente, glándulas adrenales, pudiendo producir hiperplasia de paratiroides, adenomas de hipófisis y TNP. También se puede acompañar en menor frecuencia de TCa del intestino anterior.
Epidemiología
Existen muy pocos registros de cáncer que recojan específicamente el diagnóstico de TCa y ninguno publicado que recoja los TNP.
La incidencia para los TCa es baja y muy variable según los diferentes registros, aunque parece que hay una tendencia a su incremento: el registro norteamericano del programa SEER (Surveillance, Epidemiology, and End Results) del National Cancer Institute5 recoge una incidencia de 2,3/105 en el período 1973-1991 y de 4,4/105 en 1992-1999; el registro británico6, de 1/105; el registro suizo7, de 1,9/105 en el período 1974-1985 y de 2,8/105 en 1986-1997. En una serie de autopsias realizadas sistemáticamente en Malmö (Suecia) durante un período de 12 años (1958-1969), se observó una incidencia de 8,4/ 105, que era 7 veces más alta que la incidencia recogida en el registro nacional de cáncer de todo el país8. Según el registro norteamericano, que cuenta con 13.715 casos identificados a lo largo de las últimas 5 décadas hasta 1999, en el tubo digestivo aparecen el 67,5% de los TCa, seguidos por el árbol broncopulmonar con un 25,3%. El 7,2% restante se distribuye en timo, aparato genitourinario, área otorrinolaríngea y resto de órganos internos5.
Respecto a los TNEP, se estima una incidencia de 0,4-1,2/105 a partir de datos indirectos de series publicadas9, pero no existen datos oficiales de ningún registro en el mundo.
Anatomía patológica
Según el grado de diferenciación, se distinguen 4 grupos en la más reciente clasificación de la Organización Mundial de la Salud10 (tabla 1). La mayoría de los TEGEP pertenecen a los 2 primeros grupos, es decir, tumores o carcinomas bien diferenciados. Presentan un índice mitótico y proliferativo bajo, información que tiene repercusión en el pronóstico y tratamiento. Todos ellos expresan marcadores generales neuroendocrinos como la cromogranina A, sinaptofisina, enolasa neuronal específica, CAM-5.2 o PGP-9.5 en mayor intensidad y extensión a mayor grado de diferenciación. Los tumores de origen entérico suelen expresar también otros marcadores específicos de células enterocromafines como la serotonina y, con menor frecuencia, marcadores que comparten con los TNP el como glucagón, el enteroglucagón y el polipéptido pancreático. Los TNP suelen expresar también estos marcadores generales acompañados de otros más específicos como insulina, péptido intestinal vasoactivo y gastrina11. Es importante que la muestra sea adecuada y que su obtención y procesamiento se realicen de acuerdo con protocolos estandarizados según criterios de calidad, lo que aumenta la precisión del diagnóstico12.

Presentación clínica, historia natural y pronóstico
No es infrecuente que los TEGEP se presenten con cuadros clinicorradiológicos abigarrados, con períodos largos de síntomas antes del diagnóstico, aunque otras veces éste se ve facilitado por la presencia de síntomas más específicos. La localización anatómica del tumor primario influye en su presentación clínica e historia natural y, por lo tanto, también en su potencial invasivo y pronóstico2,3. Los factores que se han relacionado de manera más concluyente con la supervivencia son el tamaño tumoral, la profundidad de la invasión local, la presencia de metástasis ganglionares, hepáticas u óseas y las características histológicas como el grado de atipia nuclear, la presencia de necrosis, el índice mitótico e índice proliferativo Ki-6713,14.
Los TCa se han clasificado tradicionalmente según su origen anatómico embrionario: intestino anterior (estómago, duodeno, páncreas, pulmón, timo), intestino medio (yeyuno, íleon, apéndice, colon ascendente) e intestino posterior (colon transverso, descendente y recto)15. Los TCa localizados en yeyuno/íleon son los más frecuentes (un 41,8% de todos ellos, seguidos en segundo lugar por los pulmonares) y el 70% presenta metástasis, aunque tienen índices de supervivencia a los 5 años que llegan al 55%. Se trata de los TCa que con mayor frecuencia (15%) pueden producir el síndrome carcinoide, cuadro clínico típico constituido característicamente por diarrea, flushing (episodios repentinos de hiperemia y rubicundez de predominio facial) y sibilancias (tabla 2), causados por la secreción de serotonina, taquicininas, bradicininas y prostaglandinas. Los TCa de recto son los segundos más frecuentes del aparato digestivo (un 27,4% de todos los TCa); suelen ser pequeños (< 1 cm), se diagnostican por colonoscopia y tienen escaso potencial invasivo (14% M1). Los metastásicos tienen una supervivencia a los 5 años del 30% y no acostumbran presentar secreción endocrina evidente. Los TCa del resto del colon son infrecuentes, pero muestran un comportamiento mucho más agresivo; hasta el 71% presenta metástasis, con una de supervivencia a los 5 años del 20%, y tampoco suelen ser funcionantes. Los TCa gástricos representan el 8,7% de todos los TCa y son los terceros más frecuentes en el aparato digestivo. Se dividen en 3 subtipos: los de tipo I se asocian a gastritis atrófica tipo A, constituyen el 75% de los casos, acostumbran ser tumores menores de 1 cm y metastatizan en un 10% de los casos; los de tipo II están asociados al síndrome de Zollinger-Ellison (un 5-10% de los casos), suelen medir menos de 1,5 cm y alrededor de un 25% metastatiza; por último, los de tipo III o esporádicos (un 15-25% de los casos) tienen un tamaño y potencial metastatizante mayores. Ocasionalmente los de tipo III pueden presentar un síndrome carcinoide atípico en forma de erupción crónica causada por histamina3,5. Los TCa apendiculares suelen ser localizados y pequeños (< 1 cm), se descubren incidentalmente en una apendicectomía, la mayoría son benignos o con escaso potencial invasivo (10% M1) y, si hay metástasis, presentan una supervivencia a los 5 años del 34%. En las series clásicas (1975) se hallaban en primer lugar en frecuencia16 y han ido en retroceso hasta ocupar sólo el 7% de todos los TCa en la actualización más reciente del registro norteamericano, por detrás de los TCa intestinales, pulmonares, rectales y gástricos5.

Los TCa pancreáticos son anecdóticos y presentan un problema de diagnóstico diferencial con los tumores neuroendocrinos de islotes pancreáticos no funcionantes. Éstos presentan habitualmente un comportamiento potencialmente invasivo, con tendencia a desarrollar metástasis a distancia, más frecuentemente ganglionares y hepáticas. Corresponden a más del 50% del grupo de TNP y pueden secretar péptidos hormonales silentes como el polipéptido pancreático (PPomas), neurotensina (neurotensinomas) o concentraciones subclínicas de otros péptidos funcionantes. Los TNP funcionantes, a excepción de los insulinomas, que en alrededor del 10% de los casos son malignos, muestran un comportamiento similar, aunque pueden diagnosticarse con un menor tamaño tumoral por presentar síntomas más evidentes. Por ello, en algunos estudios se ha postulado que tienen un comportamiento menos agresivo y mejor pronóstico que los TNP no funcionantes, observación que no se ha confirmado en otras series. Pueden ser productores de hormonas peptídicas pancreáticas y entonces dan lugar a síndromes funcionantes más o menos específicos según el predominio de una o varias hormonas (tabla 2). Por ejemplo, el síndrome de Zollinger-Ellison (gastrinoma) está mediado por gastrina y el insulinoma da lugar a una producción excesiva de insulina y proinsulina que producen cuadros de hipoglucemia. Otros TNP extremadamente raros presentan producción hormonal ectópica de corticotropina, hormona liberadora de corticotropina, hormona liberadora de hormona de crecimiento o proteína relacionada con la hormona paratiroidea3,17.
Marcadores séricos tumorales
La identificación del tipo de secreción hormonal y su cuantificación son importantes tanto para el diagnóstico como para el seguimiento. Pueden orientar sobre la localización del tumor primario en los procesos diagnósticos iniciales y, además, la monitorización de los que resultan elevados en el estudio inicial permite también complementar a las técnicas de imagen en la evaluación de la eficacia de los tratamientos. Por otra parte, cualquier tumor endocrino pancreático puede sintetizar y secretar varias hormonas, por lo que con relativa frecuencia aparecen síndromes mixtos. Asimismo, a lo largo de su evolución pueden presentar producción de hormonas no secretadas previamente1.
El diagnóstico bioquímico de los TEGEP se basa en marcadores generales y específicos. Los mejores marcadores generales son la cromogranina A y el polipéptido pancreático. La cromogranina A es una proteína secretora presente en los gránulos densos de las células neuroendocrinas. Al estar distribuida de forma ubicua en los tejidos neuroendocrinos es el marcador tumoral más sensible (60-90%), independientemente del tipo y localización tumorales, y valores iniciales muy altos obtuvieron significación pronóstica independiente en un estudio multivariado de una larga serie de TC avanzados18. En la tabla 2 se exponen los marcadores específicos responsables de los síndromes clínicos bien definidos. La determinación del 5-hidroxiindoleacético (5-HIAA) en orina de 24 h es la que se utiliza con mayor frecuencia para el diagnóstico del síndrome carcinoide. Tiene una sensibilidad mayor del 70% y una especificidad cercana al 100%, si se descartan elevaciones falsamente positivas resultantes de la ingesta de algunos alimentos o fármacos, aunque pueden observarse valores elevados también en pacientes que no presentan síntomas específicos17.
Técnicas de imagen
La tomografía computarizada (TC) de alta definición normalmente es la primera prueba de imagen donde se que puede identificar una o varias lesiones sospechosas. La TC debe realizarse siguiendo un protocolo homogéneo que incluya la inyección de contraste yodado y la evaluación completa de la fase arterial, parenquimatosa y portal, puesto que las metástasis hepáticas son muy frecuentes y pueden estar muy vascularizadas. Por otro lado, en un estudio reciente comparativo de evaluación de metástasis hepáticas la resonancia magnética tuvo una mayor sensibilidad19. La gammagrafía con octreótido es la técnica con mayor sensibilidad y especificidad para el diagnóstico y estudio de extensión de los TEGEP bien diferenciados diseminados, puesto que el 80% de ellos expresan receptores del tipo 2 de somatostatina, excepto los insulinomas, que lo expresan sólo en un 50%, lo que se relaciona con una menor sensibilidad20. La utilización racional de las técnicas de imagen durante el proceso diagnóstico ha sido objeto de un consenso cuya finalidad es mejorar la eficiencia de los procedimientos21.
Por último, para la localización de tumores primarios no aparentes con las otras técnicas, la ecoendoscopia pancreática, con o sin punción aspirativa con aguja fina, y el tránsito intestinal con cápsula endoscópica son pruebas con una buena sensibilidad, especialmente indicadas en casos con tumores primarios pequeños. La tomografía por emisión de positrones (PET) con 18 F-fluorodeoxiglucosa (FDG-PET) no es útil en los tumores bien diferenciados22 y sólo los tumores con una actividad proliferativa alta o mal diferenciados muestran un incremento en la captación de FDG. Por ello se han desarrollado nuevos trazadores con precursores de aminas marcados con 11C, de los cuales el más sensible parece ser el 5-hidroxi-L-triptófano (5-HTP). En estudios comparativos la 5-HTP-PET demostró su superioridad con respecto a la TC y la gammagrafía con octreótido23, aunque su uso está limitado porque el trazador tiene una vida media muy corta y sólo está disponible en el centro de la Universidad de Uppsala.
Protocolo diagnóstico
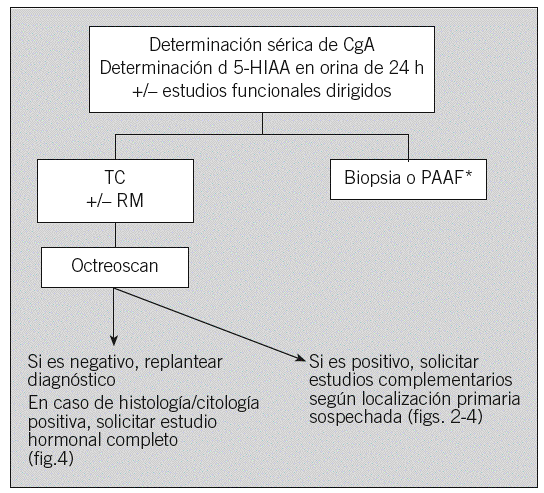
Ante la sospecha de un TEGEP (tumor ya localizado en la TC con clínica e imagen compatibles) se debe considerar la realización de un estudio hormonal inicial con cromogranina A sérica, 5-HIAA en orina de 24 h, gammagrafía con octreótido y biopsia del tumor (figs. 1-4), excepto en algunos casos de TNP localizados diagnosticados por estudios funcionales, en los que se puede considerar la resección quirúrgica sin diagnóstico histológico previo. Por ejemplo, el gastrinoma exige demostrar hipergastrinemia en ayunas y para el insulinoma la prueba diagnóstica clásica consiste en el ayuno de 24-72 h, con determinación de las concentraciones séricas seriadas de glucosa, péptido C e insulina cada 4 a 8 h, así como de proinsulina si se duda del diagnóstico. El estudio anatomopatológico debe proporcionar información sobre la morfología y el grado de diferenciación, al igual que otros factores como la inmunohistoquímica de marcadores neuroendocrinos generales y específicos y factores pronóstico como la invasividad, necrosis, índice mitótico e índice proliferativo Ki-67. En casos de enfermedad avanzada se deberá intentar obtener una biopsia lo más amplia posible y sólo en casos excepcionales se contemplará la obtención de material para diagnóstico mediante punción aspirativa con aguja fina12. En los casos que comienzan con metástasis hepáticas de origen no filiado, lo más rentable es realizar el estudio histológico y un estudio sérico hormonal completo (fig. 4).
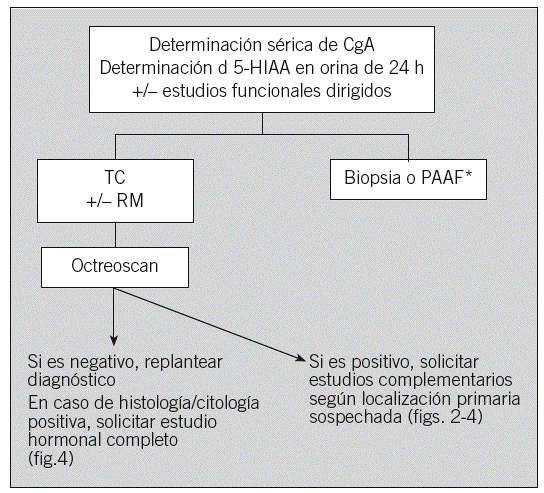
Fig. 1. Sospecha de tumor endocrinogastroenteropancreático. CgA: cromogranina A; 5-HIAA: 5-hidroxiindoleacético; TC: tomografía computarizada; RM: resonancia magnética; PAAF: punción aspirativa con aguja fina; octreoscan: gammagrafía de receptores de somatostatina. *Excepto en tumores funcionantes resecables con indicación quirúrgica.
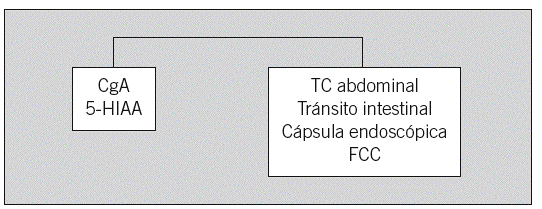
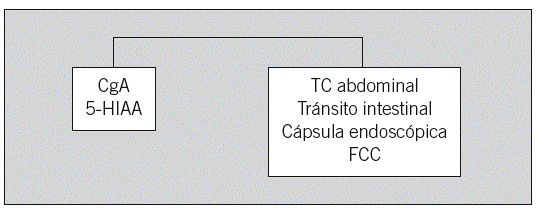
Fig. 2. Sospecha de tumor carcinoide intestinal (yeyuno, íleon, apéndice, colon proximal). CgA: cromogranina A; 5-HIAA: 5-hidroxiindoleacético; TC: tomografía computarizada; FCC: fibrocolonoscopia.
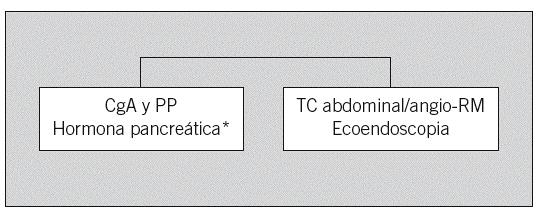
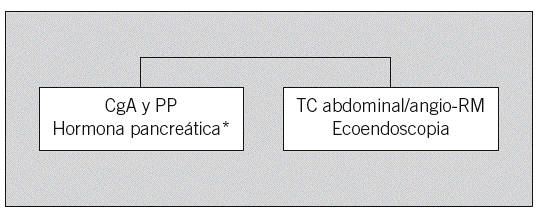
Fig. 3. Sospecha de tumor endocrino pancreático. CgA: cromogranina A; PP: polipéptido pancreático; TC: tomografía computarizada; RM: resonancia magnética. *Insulina, proinsulina, péptido C, gastrina, glucagón, somatostatina, péptido intestinal vasoactivo, corticotropina. Añadir calcio iónico/hormona paratiroidea y calcitonina/antígeno carcinoembrionario para descartar síndrome de las neoplasias endocrinas múltiples tipos 1 y 2, respectivamente.

Fig. 4. Metástasis hepáticas de origen neuroendocrino desconocido. CgA: cromogranina A; PP: polipéptido pancreático; 5-HIAA: 5-hidroxiindoleacético. *Insulina, proinsulina, péptido C, gastrina, glucagón, somatostatina, péptido intestinal vasoactivo, corticotropina. Añadir calcio iónico/hormona paratiroidea y calcitonina/antígeno carcinoembrionario para descartar síndrome de las neoplasias endocrinas múltiples tipos 1 y 2, respectivamente.
Tratamiento de la enfermedad localizada o resecable
La cirugía radical mediante resección del tumor primario y de las metástasis ganglionares o hepáticas es el único tratamiento potencialmente curativo de los TEGEP. No existe indicación de tratamiento adyuvante cuando la cirugía es radical. Incluso cuando no se consigue la curación, la cirugía puede prolongar la supervivencia, facilitar el control de síntomas relacionados con la secreción hormonal y prevenir o resolver complicaciones locales del tumor primario o de las metástasis. En los TCa resecables está indicada la exéresis oncológica, excepto en casos claros de tumores benignos como algunos TCa gástricos o en TCa de bajo potencial maligno como el de apéndice o recto de un tamaño inferior a 1 cm, en los cuales se puede recurrir a una resección local de márgenes libres como única maniobra quirúrgica24. Los TNP no funcionantes acostumbran presentarse como tumores de gran tamaño y requieren cirugía pancreática mayor con linfadenectomía regional en todos los casos25. En los TNP funcionantes el tipo de cirugía dependerá del tamaño, la localización y el potencial maligno (el 90% de los insulinomas son benignos), por lo que puede variar desde una simple enucleación hasta una pancreatectomía parcial o total. Los insulinomas y gastrinomas son los más frecuentes y en un 10-20% de los casos forman parte del síndrome de las neoplasias endocrinas múltiples tipo 1. En el contexto del síndrome de las neoplasias endocrinas múltiples tipo 1, el tratamiento de los insulinomas consiste en la enucleación de las lesiones en la cabeza del páncreas y pancreatectomía caudal cuando hay tumores localizados en cuerpo y cola del páncreas. En los gastrinomas las lesiones suelen ser pequeñas y múltiples, las recurrencias son frecuentes y el beneficio de la resección quirúrgica no está suficientemente demostrado en tumores menores de 2-3 cm. Aunque el pronóstico vital a largo plazo es bastante bueno, la causa más frecuente de muerte es el eventual desarrollo de metástasis hepáticas, lo que apoya una enfoque quirúrgico agresivo en los estadios iniciales de la enfermedad26.
Tratamiento de la enfermedad avanzada o irresecable
Los TEGEP mal diferenciados son poco frecuentes, tienen mal pronóstico y se tratan con quimioterapia paliativa basada en cisplatino27. En los grupos de TEGEP bien diferenciados, el 45% de los TCa y el 76% de los TNP presentan metástasis en el momento del diagnóstico y hasta un 20-25% más puede desarrollarlas a lo largo de su evolución. Sin embargo, debido a que suelen presentar una lenta progresión y a los avances en el tratamiento pluridisciplinario, los índices de supervivencia a los 5 años pueden llegar al 50% para los TCa y al 34% para los TNP1-3,5. En algunas ocasiones la enfermedad metastásica puede tener un curso indolente y puede mantenerse una actitud expectante, aunque el 90% de los casos progresan al cabo de un año y por ello se debe hacer un seguimiento clínico, bioquímico y radiológico para no retrasar el inicio del tratamiento, puesto que el volumen de la enfermedad diseminada es un factor pronóstico reconocido28.
Los TCa con metástasis hepáticas deben tratarse inicialmente con resección de la masa tumoral cuando pueda garantizarse un mínimo del 90% de reducción tumoral. La cirugía de metástasis hepáticas debe ser agresiva29 y en algunos casos muy seleccionados se puede incluso plantear el trasplante hepático30. Si se localiza el tumor primitivo en el intestino delgado, se debe considerar la resección quirúrgica radical del tumor incluso ante la presencia de metástasis irresecables, para anticiparse a posibles problemas de isquemia y oclusión intestinal provocados por la reacción desmoplásica típica en esta localización24. Los tratamientos ablativos como la radiofrecuencia pueden complementar la cirugía agresiva al aplicarla sobre lesiones localizadas en el remanente hepático. También puede contemplarse la radiofrecuencia en casos seleccionados con un número limitado de metástasis bilobulares irresecables31. En casos no erradicables, la embolización o quimioembolización es eficaz para el control sintomático y antiproliferativo32, y en un estudio se han demostrado una ventaja en la supervivencia libre de progresión y una clara tendencia favorable en la supervivencia global33 en combinación con el tratamiento sistémico secuencial con interferón en TCa intestinales. La combinación de interferón con análogos de somatostatina puede retrasar la progresión tumoral34 y mejorar el control del síndrome funcional35, en particular si existe positividad para receptores de somatostatina4. La quimioterapia es poco eficaz en el tratamiento de los TCa bien diferenciados con un índice proliferativo bajo36,37.
Los TNP presentan mayor sensibilidad a la quimioterapia basada en combinaciones de estreptozotocina, 5-fluorouracilo y doxorrubicina38,39, y por ello se debe valorar como primera línea en la enfermedad avanzada, incluso con intención neoadyuvante en algunos casos con el fin de facilitar una cirugía hepática más radical, la cual se indica siguiendo los mismos criterios que para los TCa y puede complementarse también con otros tratamientos ablativos y locorregionales29-31. La embolización es igual de efectiva que en los TCa y debe considerarse conjuntamente con la quimioterapia32. El interferón facilita el control del síndrome funcional refractario40 y puede tener un efecto citostático en los tumores con baja actividad proliferativa, por lo que su uso está aceptado por algunos grupos como tratamiento sistémico antitumoral de segunda línea o como alternativa a la quimioterapia en casos seleccionados35. Los análogos de somatostatina son igualmente efectivos para el control funcional y combinan bien con el resto de los tratamientos4.
Otro tratamiento cuya eficacia se ha confirmado recientemente es la administración sistémica de isótopos radiactivos unidos a análogos de somatostatina, que puede considerarse en cualquier momento del curso evolutivo de los TEGEP bien diferenciados con positividad en la gammagrafía con octreótido, aunque de momento no están disponibles en nuestro país41. Para TCa avanzados de localizaciones menos habituales el abordaje debe fundamentarse en estudios pequeños y evidencias indirectas extraídas de la bibliografía basada en los TEGEP de localizaciones más frecuentes: intestino delgado y páncreas. Se recomiendan tratamientos quirúrgicos y locorregionales siempre que sean técnicamente posibles, así como tratamiento sistémico basado en quimioterapia o interferón dependiendo del índice proliferativo y de la rapidez en la evolución, con la adición de análogos de somatostatina en los casos con síndrome funcional y con positividad en la gammagrafía con octreótido1-5.
Conclusión y perspectivas de futuro
En conclusión, los TEGEP constituyen un conjunto de entidades poco frecuentes con rasgos biológicos comunes, pero gran variabilidad en cuanto a su presentación y características clínicas, y de complejo abordaje diagnóstico y terapéutico. Diversas especialidades médicas y quirúrgicas participan habitualmente en la asistencia a los pacientes con este trastorno, que además precisa de importantes recursos de laboratorio y técnicas de imagen. Tienen un alto índice de supervivencia, a pesar de que la mayoría de los pacientes presenten enfermedad avanzada, y su manejo debe ser pluridisciplinario, con la incorporación de técnicas agresivas de resección quirúrgica de tumores localmente avanzados y metástasis hepáticas, tratamientos locorregionales como la embolización o la radiofrecuencia y avances en el tratamiento sistémico. Su incidencia en España es desconocida, así como también su impacto en cuanto a morbilidad y mortalidad. Por todo ello se ha creinos (www.getne.org), que tiene la misión fundamental de mejorar el diagnóstico y tratamiento de los pacientes con tumores neuroendocrinos en un contexto nacional, interdisciplinario y científico. Entre los proyectos activos del grupo destacan la coordinación de un registro nacional hospitalario de este tipo de tumores (www.getne.org/rgetne.htm), la edición de un manual de diagnóstico y tratamiento, establecer centros de referencia y circuitos de derivación de pacientes para técnicas complejas de diagnóstico y tratamientos, así como la realización de ensayos clínicos multicéntricos e internacionales con nuevos fármacos y la promoción de proyectos de investigación translacional.
