


The immune system constitutes a set of effective host responses to pathogens. T lymphocytes orchestrate these responses through specific receptor recognition of pathogen-derived antigenic peptides on antigen presenting cells (APC). Extracellular stimuli (such as fungal and bacterial endotoxins) and cytokines (IFN-γ, TNF-α and IL-1β) activate macrophages to express inducible nitric oxide synthase (iNOS) and inhibit pathogen replication by releasing a variety of effector molecules including nitric oxide (NO). However, the participation of NO in the resolution of immune responses is not an exclusive hallmark of iNOS from macrophages. Recent evidence indicates that constitutive NOS expression in other immune cells also plays an important role in the development of adaptive immune responses. This review summarizes recent reports indicating an important role for NO in immune responses, focusing on the unresolved controversies concerning the production and function of NO in T lymphocytes.
Los linfocitos T juegan un papel central en la regulación de la respuesta inmune adaptativa generada por el huésped frente a la agresión por agentes patógenos, mediante el reconocimiento de péptidos antigénicos expuestos en la superficie de células presentadoras de antígeno (APC). La presencia de células necróticas, la infección por virus u hongos, ciertas endotoxinas bacterianas (como por ejemplo el LPS) o las citocinas proinflamatorias IFN-γ, TNFα e IL-1β activan la expresión de la forma inducible de óxido nítrico sintetasa (iNOS) en macrófagos y con ello la producción de altas concentraciones de óxido nítrico (NO) cuya función más reconocida en estas células es mantener una actividad microbicida e inhibir la proliferación de agentes infecciosos. Sin embargo, la producción de NO y su participación en la resolución de la respuesta inmune no es una característica exclusiva de la expresión de iNOS en macrófagos. Evidencias recientes indican que la expresión de las isoformas constitutivas de NOS (cNOS: NOS endotelial y NOS neuronal) por parte de otras células inmunitarias también juega un importante papel en el desarrollo de la respuesta inmune adaptativa. La presente revisión trata de integrar los hallazgos iniciales y otros estudios más recientes en los cuales el NO ha sido considerado un mediador químico con una importante función reguladora para el desarrollo de la respuesta inmune adaptativa, centrándonos en la controversia suscitada entorno a su producción y función en los linfocitos T.
Nitric oxide (NO) is a pleiotropic and short-lived free radical, which is now recognized as an ubiquitous biomessenger existing in a wide variety of organisms. In mammals, NO has a number of pathophysiological functions. Beneficial properties of NO have been observed in the regulation of vascular relaxation, platelet aggregation, neurotransmission, cellular respiration, and the modulation of immune responses. Detrimental effects of NO include severe vasodilatation and myocardial depression during bacterial sepsis and cytotoxic tissue-damage associated with autoimmune and chronic allergic inflammation(1,2).
NO is generated by at least three different isoforms of NO synthases (NOS), which convert L-Arg and molecular oxygen to L-citrulline and NO in the presence of NADPH and tetrahydro-biopterin (BH4), a reaction that can be inhibited by analogues of L-Arg(3,4). Neuronal and endothelial NOS (nNOS/NOS1 and eNOS/NOS3, respectively) are constitutively expressed in cells as preformed proteins that are activated in response to cell-specific stimuli through the elevation of intracellular Ca2+ concentrations and the binding of calmodulin. These isoforms produce low amounts of NO as a part of normal physiological responses. In the case of eNOS, activation is positively regulated by PI3K/AKT through direct phosphorylation of Ser 1177, which increases its activity several-fold(5,6). In contrast, inducible NOS (iNOS/NOS2) is Ca2+-independent, is absent from resting cells, and is expressed in response to pro-inflammatory cytokines (e.g. IFN-γ, TNF-α and IL-1 β) and/or microbial stimuli (LPS); iNOS is characterized by pathophysiological high-output production of NO(7). NO can be converted to NO2, NO2-, NO3- and other reactive nitrogen intermediates (RNI), among which it is worth mentioning S-nitrosothiols (S-NO), peroxynitrite (ONOO-) and nitrosyl-metal complexes, which are directly implicated in the NOS-mediated post- translational regulation of proteins (S-nitrosylation of cysteines, nitration of tyrosines, and nitrosylation of prosthetic groups, respectively)(8).
iNOS has been classically described as the immunological NOS, and macrophages are the prototypic iNOS expressing cells(9–11). Even though human macrophages seem to express iNOS only under restricted circumstances(12), macrophagederived NO can act as both a highly cytotoxic molecule generated in response to microbial stimuli and proinflammatory cytokines and as a regulatory molecule that controls T lymphocyte proliferation and cytokine secretion during adaptive immune responses.
The NO produced by iNOS exerts antimicrobial actions through several mechanisms(13). Nitric oxide can inhibit bacterial replication by binding directly to double stranded DNA, causing deamination and breakage, and by disrupting zinc metalloproteins involved in DNA synthesis. Nitric oxide also impairs bacterial function, by disrupting heme-containing bacterial enzymes and oxidizing bacterial lipids. Viral infection is impaired through inhibitory NO actions on replication and the activation of proteases involved in virus entry. A more subtle action of iNOS in the regulation of innate immunity has been identified in the protective response of mice to the protozoan parasite Leishmania major(14). In this experimental model, small quantities of iNOS-derived NO, generated focally by activated macrophages, activate natural killer (NK) cells to respond to IL-12 and IFN-α/β, which allows them to become cytotoxic and to release IFN-γ, thus controlling the spread of the parasite. This initial response is critical for the containment of the infection and hence for subsequent T celldependent clinical resolution of the disease, despite the fact that in the late phase of infection iNOS is further up-regulated by IFN-γ producing CD4+ T lymphocytes and the antimicrobial activity of cytotoxic cells predominates(14,15).
Thus, even in the development of innate immune responses, NO generated by iNOS is much more than just a cytotoxic molecule with microbicidal activities in the defence against pathogens. Indeed, there is increasing evidence for an important immunoregulatory role for NO in the development of the adaptive immune responses associated with autoimmune and allergic diseases(16). The role of NO in the regulation of T lymphocyte biology is linked to some of the most important immunopathologies, including rheumatoid arthritis, asthma, diabetes, systemic lupus erythematosus, and septic shock(17). At present, there is open debate about whether NO exacerbates or reduces autoimmune and allergic chronic inflammation. NO from iNOS-expressing cells suppresses mouse T cell proliferation and ameliorates T cell-mediated murine autoimmune diseases such as graftvs-host disease(18,19), whereas proliferative and anti-proliferative effects of NO on human T cells have both been identified, through the use of L-arginine analogues and NO donors, respectively(20,21).
This review summarizes recent studies that provide insight into the role of NO in adaptive immune responses, and focuses on five areas: (i) the dual effects of exogenous NO on the immune response; (ii) the expression and possible function of NOS in T cells; (iii) the role of NO in Th1/Th2 differentiation; (iv) the generation and function of regulatory T cells; and finally (v) in thymic selection.
DUAL ACTIONS OF NO IN THE IMMUNE RESPONSEDefinition of the role of NO in inflammation and immunity presents an intractable challenge, since both protective and detrimental effects have been extensively reported(2,16,22,23). These opposing effects may ultimately reflect the dual actions of NO on a broad spectrum of cell processes(24). The use of NO donors, NOS inhibitors and the study of experimental models of inflammation in NOS-deficient mice have provided solid evidence that NO is able to promote either suppression or activation(21,25,26), death or survival(13,27), and gene transcription or repression(28,29). In all these cases, the outcome depends on the enzymatic and cellular sources of NO, the priming of immune cells, the biological redox milieu, and the effective concentration of NO, which itself frequently depends on the distance of the target cell from the source(30). These properties of NO may explain the difficulties in determining its exact role under pathophysiological conditions.
The immunosuppressive actions of NO in the response to infection are well-established, and are not necessarily dependent on its anti-proliferative effects on T cells. For instance, bacterial superantigens are potent T cell activators that induce active cell proliferation initially, but also induce cell death at later stages(31). Inhibition of iNOS prolongs the enteropathy induced in mice by superantigen staphylococcal enterotoxin B (SEB) and also increases morbidity and mortality by up-regulating IFN-γ and TNF-a production(26,32,33), indicating that high levels of NO are important for recovery from superantigen-induced septic shock. Furthermore, T celldependent immunosuppression is also mediated by specialized myeloid suppressor cells (MSCs), which also produce NO(25). MSCs express Gr-1, the β2 integrin Mac1, iNOS, and arginase, which converts L-Arg into L-ornithine and urea. MSCs are frequently found in advanced mouse tumours, and have been proposed to promote the immunological escape from tumour infiltrating lymphocytes, affecting the metabolism of L-arginine in T cells(34). In MSCs, the coordinated activities of arginase and a plasma membrane carrier of positively charged amino-acids deplete the extra-cellular milieu of LArg, and this results in down-regulation of TCR CD3ζ expression and subsequent arrest of T lymphocyte proliferation(35). MSC-mediated suppression of T cell proliferation may involve synergy between iNOS and argininase, since when both enzymes are activated, the depletion of L-Arg will stimulate iNOS to produce traces of superoxide (O2−), which reacts with NO to produce peroxinitrite (ONOO−) and other RNIs capable of inducing T lymphocyte apoptosis(25). Another mechanism by which iNOS-derived NO might regulate immunosuppression is suggested by the observation that high concentrations of NO impair IL-2R-induced signaling, thereby blocking the activation of Janus kinases (JAK1 and −2) and the signal transducer and activator of transcription factor 5 (STAT5)(18,36).
Although the concentration threshold above which NO induces apoptosis varies considerably among cells, in general, high concentrations of NO induce apoptosis in T cells whereas low concentrations have anti-apoptotic effects. Apoptotic-inducing mechanisms are activated in mitochondria through the intrinsic pathway or are triggered by death receptors such as CD95/Fas, a member of the tumor necrosis factor receptor family. Fas ligand (CD95L/FasL) interacts with the Fas-associated death domain (FADD) and recruits pro-caspase-8, which is proteolytically activated and released to the cytosol(37).
In mitochondria, NO competes with O2 for binding to cytochrome C oxidase of the mitochondrial electron transport chain. The resulting reduction in mitochondrial membrane potential leads to the generation of RNI that nitrosylate cytocrome c on its heme iron, triggering its release into the cytosol(13). The realeased cytochrome c associates with the adapter protease activating factor 1 (Apaf-1) to assemble the apoptosome, a multimeric complex that induces cell death by recruiting and activating caspases 2, 3, 7, and 9.
There is also evidence that NO mediates apoptosis through other pathways. Experiments in thymocytes from p53-null mice or human lymphocytes mutated on p53 suggest that these cells are more resistant to NO-induced apoptosis, supporting the idea that p53 may transmit pro-apoptotic responses induced by NO(38,39). Regarding this mechanism, it has been suggested that NO retains p53 in the nucleus by increasing p53 phosphorylation and ubiquitination through the regulation of the p38 and c-Jun N-terminal kinase (JNK) mitogen-activated protein kinases (MAPK). NO can also induce T cell apoptosis by regulating the expression of Bcl-2 members (e.g. Bcl-2, Bcl-x, and Bax) and apoptosis inducing factor (AIF), or through the S-nitrosylation-dependent nuclear translocation and degradation of glyceraldehide-3-phosphate dehydrogenase (GAPDH)(40,41).
In contrast, low-to-moderate concentrations of NO have been shown to protect T cells from apoptosis(42,43). Inhibition of constitutive NOS with Arg analogues or by substrate depletion increases apoptosis, and exogenous NO substantially represses apoptosis in T cells activated with anti-Fas antibodies. Caspases are the proposed targets through which NO exerts its anti-apoptotic effects. NO prevents caspase activation through cGMP-dependent and independent mechanisms(42). Among the cGMP independent mechanisms S-nitrosylation appears to be important(44–46). All caspases contain an essential cysteine within their active centers that is susceptible to S-nitrosylation, and S-nitrosylation of caspase−1, −3, and −9 correlates with their enzymatic inhibition in studies carried out in vitro(43,45). However, rather than a direct inhibition of caspase activities, the anti-apoptotic effect of caspase S-nitrosylation might involve an inhibition of apoptosome assembly(27,47). For example, S-nitrosylation of caspase-3 on a Cys residue away from the active site induces its association with acid sphingomyelinase (ASM), which inhibits caspase-3 proteolytic cleavage, thus protecting cells from apoptosis(48). Studies carried out in Jurkat T cells suggest that NO, rather than inhibiting caspase-3 enzymatic activity, attenuates its proteolysis to the active form, through the mitochondrial pathway, by interfering with Apaf-1/caspase-9 apoptosome assembly, and through the CD95 death receptor pathway, by S-nitrosylation of caspase-1 and −8(27,43). Nevertheless, in some cell systems, caspase inhibition alone is not sufficient to fully rescue cells. In this regard, cytokine-induced iNOS or NO donors prevent the decrease in Bcl-2 expression in splenic B lymphocytes(49), inhibit ceramide accumulation in CD95-stimulated human γδT lymphocytes(50), prevent AP-1-dependent CD95L transactivation in response to cytotoxic stress(46), inhibit apoptosis signal-regulating kinase 1 (ASK-1)(51), and diminish intracellular ATP, which may affect caspase activation by preventing cytochrome C oxidase release from mitochondria and/or the formation of the apoptosome complex(52). Temporary impairment of ATP generation, together with the production of low levels of ROS in mitochondria, may activate glycolytic enzymes and redox-sensitive transcription factors involved in anti-apoptotic effects and cytoprotection(53). However, inhibition of ATP generation in Jurkat T cells below a certain threshold may switch cells from the prevention of apoptosis to necrosis(54,55).
As mentioned above, the anti-or pro-apoptotic effects of NO probably involve specific interactions with reactive oxygen intermediates generated by the redox state of the cell. Thus, different RNI may have specific biochemical targets and elicit different biological responses, or indeed may produce opposite outcomes from different effects on the same target. An illustrative example of how RNI may coordinately regulate apoptotic cell death is the Snitrosylation/denitrosylation switch of caspase-3 and the NO-dependent expression of p53. Caspase-3 is constitutively S-nitrosylated and inactive in the mitochondria of Jurkat T cells and is activated by denitrosylation induced by proapoptotic stimuli(56). Low levels of NO from constitutive NOS would S-nitrosylate caspases and thereby inhibit apoptosis; however, in situations where there is elevated production of O2−this could react with NO to form peroxynitrite, which damages DNA leading to p53 upregulation and stimulation of apoptosis(38,57,58).
It has been reported that NO can either inhibit or increase the expression of pro-inflammatory cytokines, and that these effects are exerted by the regulation of signaling cascades and transcription factors. A large number of signaling pathways have been shown to be regulated by NO. The interaction of NO with components of the Janus-STAT cascade or with MAP kinases (e.g. extracellular signalregulated kinase ERK, JNK, and stress-activated protein kinase SAPK) have been studied in vitro(59). S-nitrosylation of cysteines in these proteins may lead to their activation or inactivation(23). Nitric oxide also modulates nuclear factor (NF)-κB. In cell free systems, NO inhibits the DNA-binding activity of NF-κB through S-nitrosylation of a crucial Cys in the p50 subunit. However, in T cells NO-dependent Snitrosylation can influence the activity of NF-κB both positively, through the activation of p21 Ras, or negatively, by preventing the degradation of inhibitory (I)- κB and/or the activity of I-κB kinase (IKK)(60–62).
In addition to pro-inflammatory cytokines, NF-κB also regulates the transcription of endothelial adhesion molecules such as vascular cell adhesion molecule 1 (VCAM-1), intracellular adhesion molecule 1 (ICAM-1), and E- and P- selectins(63), all of which are implicated in the mechanism of leukocyte transmigration towards inflammatory foci. It is well established that, under pathophysiological conditions, constitutive production of NO controls blood homeostasis and inhibits platelet and leukocyte adhesion to endothelial cells. Intravital microscopy studies and in vitro experiments using vascular perfusion or flow chambers clearly indicate that both endogenous NO and NO released from donors impede the rolling, firm adherence and transmigration of leukocytes, and reduce the expression of adhesion molecules on endothelial cells(64). However, whether the role of iNOSderived NO in modulating vascular permeability is dependent or independent of its action on adhesion molecule expression remains an open question. The expression of adhesion molecules in iNOS deficient mice does not change between basal conditions and sepsis, suggesting that iNOS may modulate leukocyte-endothelial cell interactions through effects on leukocyte function(65).
NO can also either promote or suppress the function of activating protein (AP)-1, a heterodimeric transcription factor consisting of c-Fos and c-Jun. AP-1 binds to the promoters of many genes encoding for pro-inflammatory cytokines and adhesion molecules. In cell free systems, NO inhibits DNA-binding of c-Jun and c-Fos through the Snitrosylation and glutathionylation of specific Cys residues(66,67). However, in Jurkat cells AP-1 DNA-binding in response to CD95-induced apoptotic signals is inhibited by NO donors, whereas in primary lymphocytes nuclear accumulation of AP-1 in response to TNF-α and IL-1β is blocked by NOS inhibitors, and peroxynitrite induces the translocation of AP-1 to the nucleus(46,68). It is significant that the iNOS gene promoter itself contains NF-κB and AP-1 response elements and that these transcription factors are important for iNOS gene expression(69). Through this route, NO from constitutive eNOS is able to regulate iNOS expression during septic shock, which indicates that eNOS, aside from its role in vascular homeostasis, has proinflammatory functions(70).
Another group of transcription factors with important roles in the control and activation of T lymphocytes is the nuclear factor of activated T cells (NF-AT) family. NO inhibits NF-AT translocation to the nucleus in allo-stimulated T lymphocytes(71), presumably via a cGMP-dependent mechanism since in cardiac myocytes this effect appears to be mediated by the stimulation of soluble guanylate cyclase (sGC)(72).
NOS EXPRESSION AND FUNCTION IN T CELLSExpression of NOS and production of NO are characteristic of cells involved in immune responses, and the following cell types are important sources of NO: NK cells, eosinophils, mastocytes, neutrophils, dendritic cells (DCs), macrophages, and tissue-specific cell types associated with host defence (endothelial cells, epithelial cells, myocytes, fibroblasts, keratinocytes, hepatocytes and glial cells)(2). Both iNOS and eNOS expression have been reported in macrophages, DCs and NK cells(2), whereas only iNOS expression has been detected in Epstein-Barr and Burkitfs virus-infected lymphoma B cell lines(73). Although probably every type of mammalian cell is capable of generating NO, whether or not T lymphocytes express any NOS isoform and produce NO during the immune response has been a matter of intense debate.
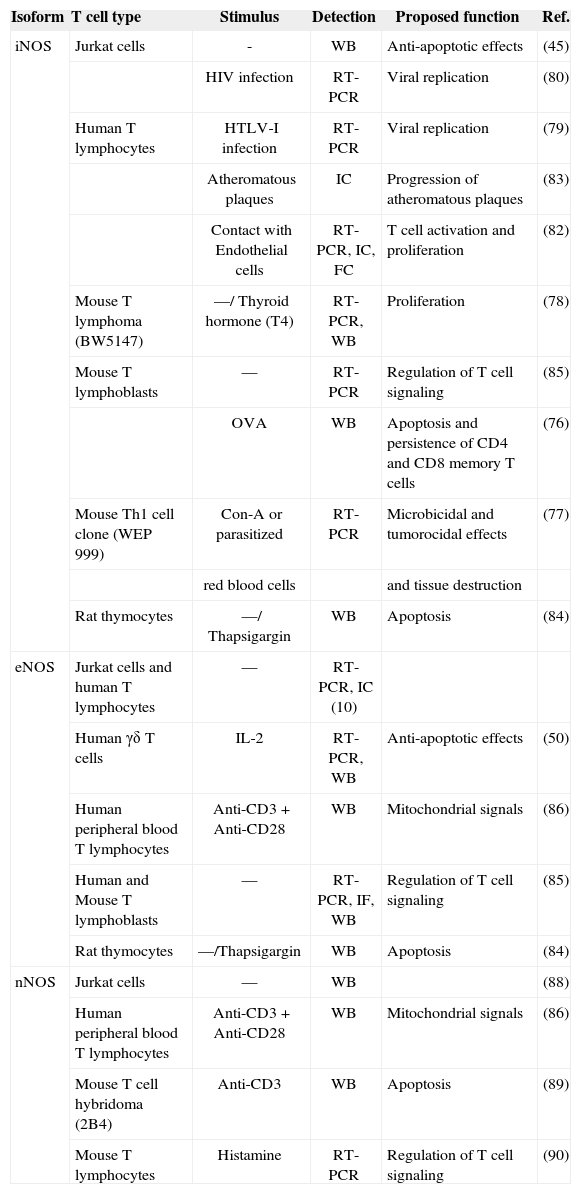
The control of T lymphocyte function and proliferation is crucial to the homeostatic balance that exists in the immune system. Loss of this balance, leading to uncontrolled proliferation or excessive reduction of effector T lymphocytes, underlies autoimmune and immunodeficiency diseases. Cell homeostasis is coordinately regulated by intra-and extracellular messengers. The release from macrophages of high levels of iNOS-derived NO induces apoptotic cell death in a variety of cells, and has been proposed to alter the responsiveness of T lymphocytes to antigen or mitogenic stimuli. However, T lymphocytes themselves also produce NO: several reports, using electrochemical detection of NO gas, NOS inhibitors, or NO fluorescent detection probes, have confirmed endogenous NO production by T cells in response to mitogens, chemoattractants and apoptosisinducing agents (Figure 1)(21,74,75). Other reports have identified mRNA and protein expression of specific NOS isoenzymes in T cells (Table I).
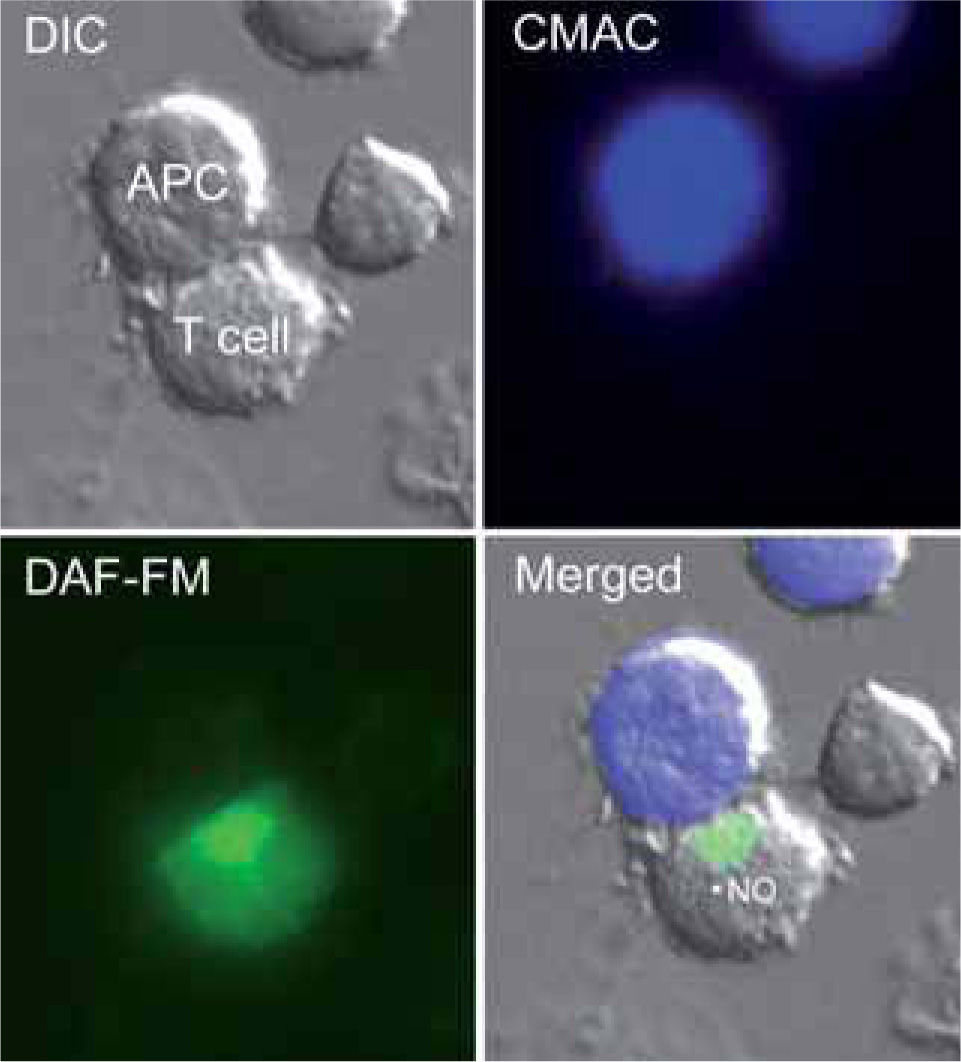
. NO production is revealed by the green fluorescence. The merged image shows fluorescence staining superposed on the DIC image.")
T cells produce NO in response to antigen-pulsed APCs.
The figure shows a Jurkat T cell loaded with the NO probe DAF-FM diacetate forming a conjugate with a superantigen E-pulsed Raji APC (stained by loading with CMAC, blue). NO production is revealed by the green fluorescence. The merged image shows fluorescence staining superposed on the DIC image.
Expression and function of NOS isoforms in human and mouse T cells
| Isoform | T cell type | Stimulus | Detection | Proposed function | Ref. |
| iNOS | Jurkat cells | - | WB | Anti-apoptotic effects | (45) |
| HIV infection | RT-PCR | Viral replication | (80) | ||
| Human T lymphocytes | HTLV-I infection | RT-PCR | Viral replication | (79) | |
| Atheromatous plaques | IC | Progression of atheromatous plaques | (83) | ||
| Contact with Endothelial cells | RT-PCR, IC, FC | T cell activation and proliferation | (82) | ||
| Mouse T lymphoma (BW5147) | —/ Thyroid hormone (T4) | RT-PCR, WB | Proliferation | (78) | |
| Mouse T lymphoblasts | — | RT-PCR | Regulation of T cell signaling | (85) | |
| OVA | WB | Apoptosis and persistence of CD4 and CD8 memory T cells | (76) | ||
| Mouse Th1 cell clone (WEP 999) | Con-A or parasitized | RT-PCR | Microbicidal and tumorocidal effects | (77) | |
| red blood cells | and tissue destruction | ||||
| Rat thymocytes | —/ Thapsigargin | WB | Apoptosis | (84) | |
| eNOS | Jurkat cells and human T lymphocytes | — | RT-PCR, IC (10) | ||
| Human γδ T cells | IL-2 | RT-PCR, WB | Anti-apoptotic effects | (50) | |
| Human peripheral blood T lymphocytes | Anti-CD3 + Anti-CD28 | WB | Mitochondrial signals | (86) | |
| Human and Mouse T lymphoblasts | — | RT-PCR, IF, WB | Regulation of T cell signaling | (85) | |
| Rat thymocytes | —/Thapsigargin | WB | Apoptosis | (84) | |
| nNOS | Jurkat cells | — | WB | (88) | |
| Human peripheral blood T lymphocytes | Anti-CD3+ Anti-CD28 | WB | Mitochondrial signals | (86) | |
| Mouse T cell hybridoma (2B4) | Anti-CD3 | WB | Apoptosis | (89) | |
| Mouse T lymphocytes | Histamine | RT-PCR | Regulation of T cell signaling | (90) |
WB: Western Blot; FC: Flow cytometry; IC: Immunocytochemistry; IF: Immunofluorescence.
Mouse T cell lines and primary T lymphocytes express iNOS and produce NO in response to TCR-mediated signals. In these cells, iNOS-derived NO reduces cytokine secretion and induces apoptosis to down-regulate potentially deleterious immune responses, while regulating the lifespan of memory T cells(76,77). It has also been reported that thyroid hormones induce iNOS expression in murine tumor T lymphocytes, increasing tumor and mitogen-induced cell proliferation(78). In contrast, although iNOS has been detected in some tumoral T cell clones(45), human T cell lines and primary T lymphocytes hardly express iNOS unless they were infected with human T-lymphotropic viruses(79,80). Nevertheless, iNOS expression has been detected by immunohistochemistry of human T lymphocytes infiltrating human arterial transplantations in immunodeficient mice(81). By using the iNOS inhibitor 1400W, the authors of this report showed that iNOS expression and high production of NO in human arterial grafts impairs contractility and results in desensitization of vascular smooth muscle cells (VSMCs). Moreover, iNOS expression and VSMC dysfunction in the arterial grafts could be prevented by inhibition of IFN-γ. This study thus suggests that IFN-γ is a central mediator of allogeneic T cell-induced dysfunction and arteriosclerosis of transplanted human allografts operating through the up-regulation of iNOS in T cells. Although the human T lymphocytes in this experimental model would express iNOS in response to murine host environmental factors, recent work shows that endothelial cells induce the expression of iNOS in human alloreactive T cells by an as yet unknown antigen-independent mechanism(82). These results corroborate previous immunochemistry studies that proposed a role for iNOS expressing T cells in atheromatous plaque progression(83). Thus, signals that regulate the induction of iNOS expression in human T lymphocytes may be species specific, induced in response to virus infection or activated in atherosclerotic processes.
Although human T lymphocytes can express iNOS, the rapid onset of low-level NO synthesis during TCR-mediated stimulation suggests the action of constitutive rather than inducible NOS(21). Initial studies reported that human T cell lines and primary T lymphocytes express eNOS mRNA(10). Moreover, γδ T lymphocytes (a subset of intraepithelial T cells involved in the first line of defence against pathogens) generate NO endogenously from eNOS. This, protects cells, through a cGMP-dependent mechanism, from CD95-tiggered apoptosis but not from other pro-apoptotic stimuli(50). Expression of eNOS has also been reported in the endoplasmic reticulum of rat thymocytes, and appears to play an important role in mitochondrial Ca2+-induced apoptosis(84). Low levels of eNOS-derived NO are synthesized in antigen-stimulated Jurkat cells and primary human and mouse T lymphoblasts during antigen-specific T cell interactions with antigen presenting cells (APC). This eNOS production plays an important role in the organization of the immune synapse and in early and late T cell activation events: increasing CD3ζ, ZAP-70 and ERK phosphorylation, and IFN-γ production, but reducing IL-2 synthesis(85). Furthermore, it has been reported that CD3-CD28 co-stimulation in T cells is associated with up-regulation of eNOS and nNOS, NO production, and elevation of the mitochondrial transmembrane potential (Aψm), resulting in mitochondrial hyperpolarization (MHP)(86). Innate and adaptive immune responses depend on the controlled production of ATP and reactive oxygen intermediates (ROIs) in mitochondria. The clonal expansion of T cells in response to antigenic-stimulation is tightly regulated, and potentially auto-reactive cells are eliminated by apoptosis. MHP appears to be the initiator step of several apoptotic pathways. Persistent MHP has been linked to increased mitochondrial mass in T lymphocytes from patients with systemic lupus erythematosus (SLE), even though exogenous rather than endogenous production of NO seems to be responsible for mitochondrial dysfunction and necrosis in these cells(87).
Expression and activity of nNOS has also been reported in human and mouse T cells(86,88). The mouse T cell hybridoma 2B4 upregulates nNOS expression upon TCR engagement and synthesizes NO in a Ca2+-dependent manner, enhancing FasL-mediated apoptosis(89). Interestingly, histamine deficiency preferentially increases nNOS expression and NO production in mouse T lymphocytes from spleen, suggesting that histamine negatively regulates NO production in activated T lymphocytes(90). Histamine selectively increases the production of Th2 cytokines and inhibits Th1 differentiation(91). Recent studies have suggested that P-selectin and P-selectin glycoprotein ligand 1 (PSGL-1) mediate recruitment of Th1 but not Th2 cells into inflamed tissues(92). Given that Pselectin-PSGL-1 interaction can be positively regulated in the presence of NO, it is reasonable to assume that histamineinduced down-regulation of nNOS-derived NO will alter the accumulation of Th1 but not Th2 cells at inflammatory foci. More recently, a specific role for NO in Th1 cell adhesion and transmigration has been reported. IFN-γ induces a potent anti-adhesive effect on Th1 cells (but not Th2 cells), reducing their recruitment to inflammatory foci in vivo(93). This impaired Th1 cell migration is not observed in mice deficient in iNOS or treated with the iNOS specific inhibitor L-NIL. Moreover, iNOS-deficient Th1 cells have a decreased ability to bind ICAM-1, despite the fact that neither endothelial cells nor Th1 lymphocytes show alterations in ICAM-1 expression. This therefore suggests that IFN-γ-dependent iNOS expression reduces the ability of Th1 cells to establish interactions with the endothelium via the ICAM-1-LFA-1 adhesion pathway.
NO AND TH1/TH2 CELL DIFFERENTIATIONNaïve CD4+ T lymphocytes recognize antigens on APC in peripheral lymphoid organs, and this results in the expansion of antigen-specific lymphocytes and their subsequent differentiation into subsets of effector cells that can be distinguished on the basis of the cytokines they secrete. Chronic inflammatory diseases are often dominated by Th1, Th2, or the recently characterized IL-17-producing T helper (Th17) cells(94). The most important differentiation-inducing stimuli for Th1 and Th2 cells are IL-12 and IL-4, respectively. Th1 cells produce IFN-γ, TNF-α, and IL-2, and their principal function is to activate cellular immunity, whereas Th2 cells typically synthesize IL-4, IL-5, IL-6, and IL-10, and regulate humoral immune responses. The regulation of cytokines by NO has received considerable attention during recent years because it might be of relevance in the selection of Th responses and the role they play in chronic inflammation.
Experiments carried out in Leishmania major-infected iNOS-deficient mice clearly indicate that high NO concentrations promote Th2 differentiation via the suppression of IL-12 synthesis by activated macrophages(95,96). Moreover, the incidence and severity of arthritis is higher in iNOSdeficient mice than in heterozygous or wild-type animals(97). Interestingly, iNOS-deficient mice also show an exacerbated response to experimental autoimmune uveitis (EAU) and encephalomyelitis (EAE). In both experimental models, administration of IL-12 to wild-type animals protects them from Th1 responses by inducing IFN-γ over-production and the consequent expression of iNOS and synthesis of NO, which confers protection by apoptotic deletion of Th1 cells(98). It thus seems likely that high levels of NO generated by iNOS prevent over-expansion of Th1 cells, which have been implicated in a variety of autoimmune diseases. In contrast, low concentrations of NO stimulate T cells to express IL-12R and promote Th1 differentiation(99). The mechanism by which low doses of NO promote the differentiation of Th1 but not Th2 cells involves the selective induction of IL12-Rβ2 via cGMP. The levels of cGMP in CD4+ T cells increase after exposition to nanomolar concentrations of NO from NO-donors, and correlate well with enhanced Th1 cell activation. NO produced by APCs activates the soluble isoform of sGC and leads to increased cGMP production and IL12-Rβ2 expression, which facilitates the differentiation of Th1 cells by IL-12-mediated signaling and TCR engagement. Evidence from in vitro studies shows that NO selectively enhances the stimulation of Th2 cells but not Th1 cells, and that the expression of iNOS is induced by the Th1 cytokine IFN-γ and inhibited by the Th2 cytokines IL-4 and IL-10, suggesting overall that NO would promote Th2 differentiation(17). Moreover, NO inhibits the production of IL-2 and increases the production of IL-4 by effector T cells, but has no effect on the secretion of IL-10(100). Since Th1 cells are more susceptible than Th2 cells to apoptosis, it seems likely that NO regulates the balance between Th1 and Th2 populations by promoting Th1 apoptosis at high concentrations and suppressing it at low concentrations(47,101). However, other studies indicate that NO donors inhibit the proliferation of Th1 and Th2 populations. The NO donors SIN-1 and SNAP inhibit the proliferation of anti-CD3 or mitogen activated human peripheral Th1 and Th2 cell clones, and prevent their release of IFN-γ, IL-2, IL-4, IL-5 and IL-10(102). It has also been reported that addition of NO to human bronchial epithelial cells reversibly suppresses the proliferation of activated Th1 and Th2 CD4+ T cells in atopic asthma(103). Thus, NO emerges as an additional signal for the regulation of subpopulations of T lymphocytes (Figure 2).
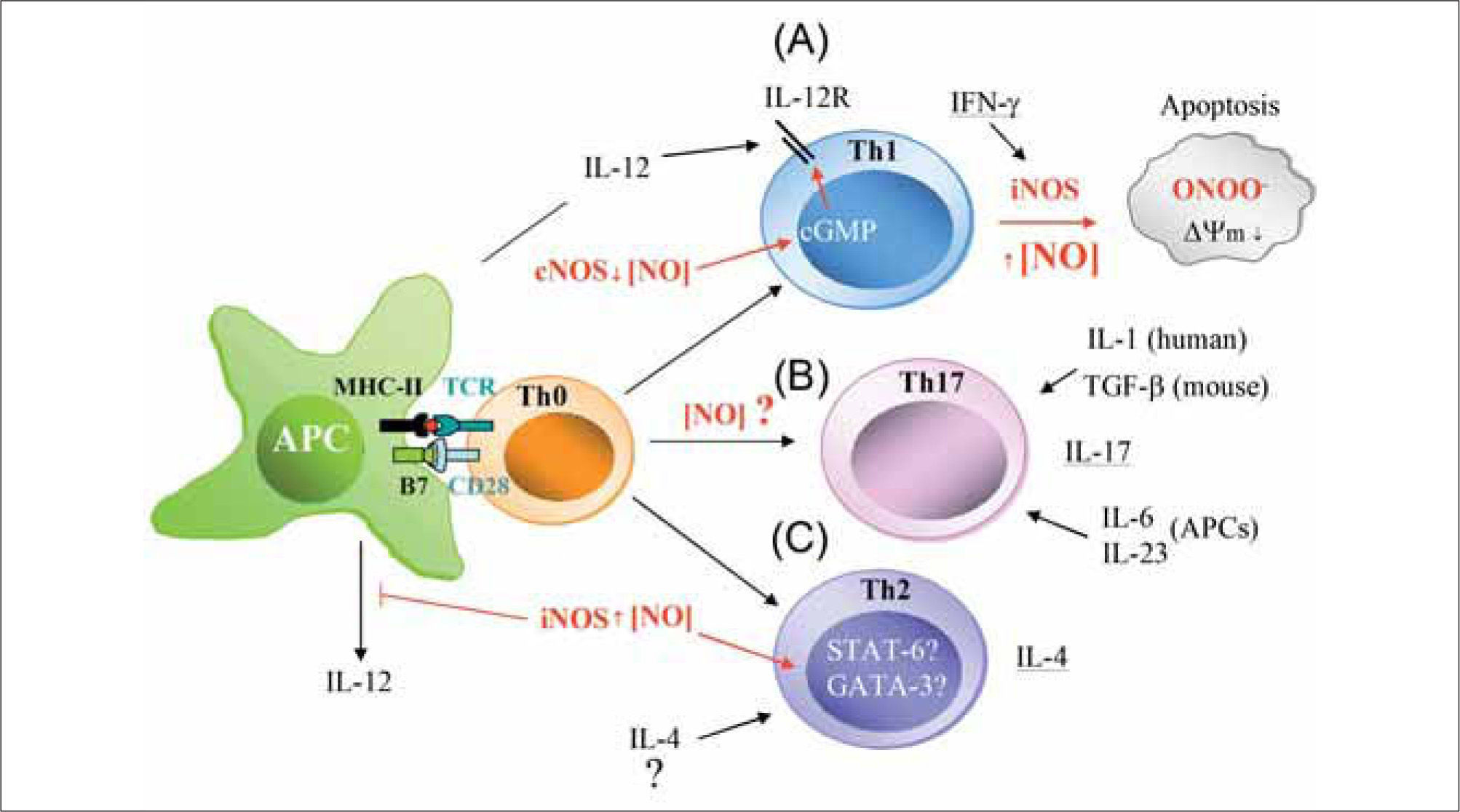
 Low levels of NO presumably from constitutive NOS (nNOS and/or eNOS) positively regúlate IL-12-mediated Th1 differentiation by increasing IL-12 receptor (IL-12R) expression on T cells through a cGMP-dependent mechanism. In contrast, large amounts of IFN-γ from exacerbated Th1 responses increase iNOS expression in immune cells. This high production of iNOS-derived NO reduces mitochondrial membrane potential (¿ψm), generates ROS (O2-), and promotes the synthesis of peroxynitrite (ONOO-), which can initiate apoptosis via Tyr nitration of proteins. (B) NO may cooperate with IL-6, IL-23, and IL-1 (human) or TGF-b (mouse) to regulate Th17 differentiation. (C) High levels of NO from iNOS positively regulate IL-4-mediated Th2 cell differentiation. The hypothetical actions of high production of iNOS-derived NO on the activation of the Th2 specific transcription factors STAT-6 and GATA-3 are indicated.")
Possible actions of NO on T cell differentiation during antigen-specific T cell-APC interactions. (A) Low levels of NO presumably from constitutive NOS (nNOS and/or eNOS) positively regúlate IL-12-mediated Th1 differentiation by increasing IL-12 receptor (IL-12R) expression on T cells through a cGMP-dependent mechanism. In contrast, large amounts of IFN-γ from exacerbated Th1 responses increase iNOS expression in immune cells. This high production of iNOS-derived NO reduces mitochondrial membrane potential (¿ψm), generates ROS (O2-), and promotes the synthesis of peroxynitrite (ONOO-), which can initiate apoptosis via Tyr nitration of proteins. (B) NO may cooperate with IL-6, IL-23, and IL-1 (human) or TGF-b (mouse) to regulate Th17 differentiation. (C) High levels of NO from iNOS positively regulate IL-4-mediated Th2 cell differentiation. The hypothetical actions of high production of iNOS-derived NO on the activation of the Th2 specific transcription factors STAT-6 and GATA-3 are indicated.
At the transcriptional level, the differentiation of naive CD4+ T cells into Th1 effectors is regulated by the transcription factors STAT-4 and T-bet, which regulate the expression of IFN-γ whereas IL-4-mediated differentiation into Th2 cells involves the activation of Jak1 and Jak3 tyrosine-kinases and the transcription factors STAT-6 and GATA-3, which trigger gene transcription of IL-4 and IL-13(104). It is well known that Ras/MAPK signaling also plays an important role in T cell differentiation: strong signals from the antigenengaged TCR sustain ERK activation and Th1 differentiation, whereas weak and transient TCR-mediated activation of ERKs preferentially induces Th2 differentiation, as a consequence of increased Jak-1 kinase activity, STAT-6 phosphorylation and GATA-3 activation(105). A role for NO in Ras/ERK-mediated Th1/Th2 differentiation is supported by the regulation of Ras and ERK activity by NO donors, via a cGMP-independent mechanism(106).
NO AND REGULATORY T CELLSSeveral types of regulatory T cells (Tregs) have been implicated in the generation of tolerance to exacerbated inflammatory responses. These include natural CD4+CD25+ T cells and adaptive Th3 and Tr1 Tregs. Natural Tregs develop in the thymus and give rise to a population of selfantigen-specific T cells in the periphery that protects from potential autoimmune damage. Adaptive Tregs can be generated by repetitive antigen presentation to naïve CD4+CD25- T cells by immature or tolerogenic DCs(107–109).
Several lines of evidence suggest an important role for NO in the generation and function of Tregs (Figure 3). First, CD4+CD25+/Foxp3+ adaptive Tregs can produce IFN-γ and interact directly with APC to inhibit antigen-driven T cell proliferation through a mechanism dependent on cell-tocell contact(110,111). The production of IFN-γ by Tregs can influence the activity of APCs in several ways. For example, suppression of T cell proliferation by mouse macrophages may depend on IFN-γ-induced iNOS expression, since IFN-γ is the main inducer of iNOS in these cells(2,112). Also, it has been reported that IFN-γ neutralization reduces the expression of iNOS mRNA in rejected skin allografts(113,114). Second, DCs expressing a kinase-defective dominant-negative form of Ikappa B kinase-2 (dnIKK2) show an impaired allostimulatory ability to upregulate MHCII antigens and costimulatory molecules in response to either lipopolysaccharide or CD40 engagement(115). Naïve mouse T cells stimulated with antigen-loaded immature dnIKK2 DCs differentiate into CD4+CD25- Tregs (dnIKK2-Tregs). These Tregs are Foxp3+, express iNOS, and have the ability to inhibit naive and pre-activated T cell responses in vitro by releasing soluble factors(115). NOS inhibitors significantly inhibit the suppressive activity of dnIKK-Tregs in a naïve mixed lymphocyte reaction (MLR). A third line of evidence for a role for NO in Treg development and function comes from studies carried out in NR206 cells, a CD4+CD25+/Foxp3- Treg cell population isolated from diabetes resistant NOR mice. In response to auto-antigens these cells secrete IFN-γ and induce NO production by APCs as a response to IL-2 produced by other activated T cells. The APC-derived NO suppresses the proliferation of pathogenic T cells, thereby inhibiting the course of autoimmune diseases. This mechanism of immunosuppression occurs only in the presence of NR206 Treg cells and large quantities of IL-2 and IFN-γ from established Th1 responses(116). Finally, recent studies have demonstrated the existence of a population of Tregs (NOTreg) whose generation depends on an NO action via p53-mediated expression of anti-apoptotic genes and local secretion of IL-2(117). NO-Tregs are a subset of T lymphocytes induced by NO together with TcR-mediated activation. They are CD4+CD25+, Foxp3-, GITR+ and CD27+, and have a Th2 phenotype suppressive against CD4+CD25-effector cells. In contrast to the more established Treg subsets (natural and adaptive Tregs), NO-Tregs do not express Foxp3. NO-Tregs also differ from Th3 suppressive cells in not producing TGF-β . However, NO-Tregs share some characteristics with Tr1 cells since the suppressive activities of both are IL-10-dependent (although, unlike Tr1 cells, the induction of NO-Tregs is IL-10-independent). The role of NO in the generation of NO-Tregs has also been demonstrated in vivo with the general NOS inhibitor L-NMMA. Based on these diverse lines of evidence, it would appear that T lymphocyte-derived NO, through a variety of mechanisms, plays a central role in regulating the strength of chronic inflammatory responses associated with autoimmune and allergic diseases.
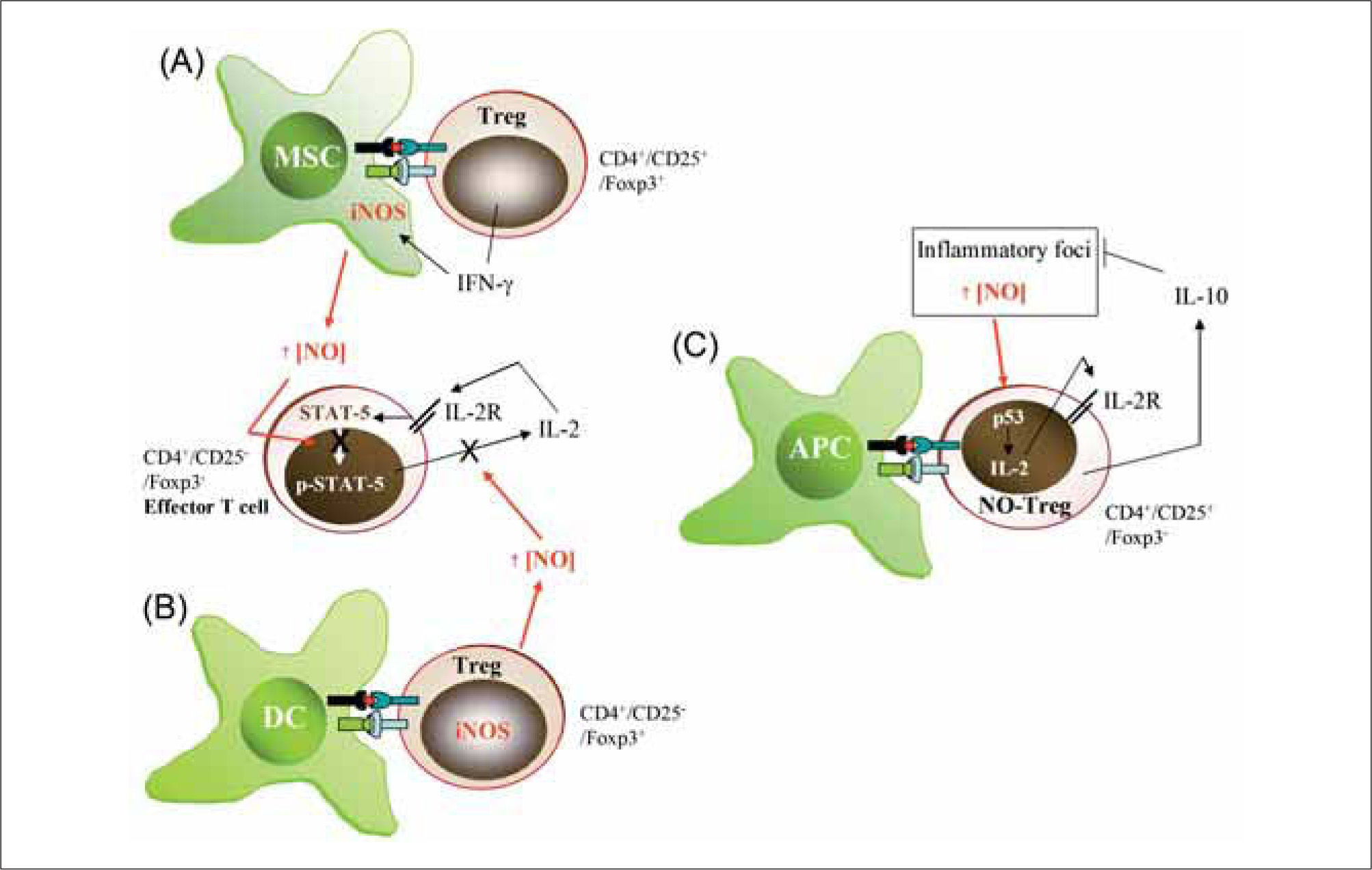
 IFN-γ producing CD4+/CD25+/Foxp3+ Tregs induce iNOS expression in myeloid suppressor cells (MSC). (B) CD4+/CD25-/Foxp3+ Tregs express iNOS and release large amounts of NO. NO from either MSC (A) or iNOS-expressing Tregs (B) is able to modulate the effector activity of CD4+/CD25-IFoxp3Th1 cells by reducing IL-2R signaling (STAT-5 activity) and IL-2 production. (C) Generation and activity of NO-Tregs. Large amounts of NO from inflammatory foci generate CD4+/CD25+/Foxp3-T cells, which activate p53 to increase IL2 production and proliferation. Similarly to CD4+/CD25+/Foxp3+ Tregs, NO-Tregs exert suppressive functions mainly through the production of IL-10.")
Influence of NO on Treg generation and function. (A) IFN-γ producing CD4+/CD25+/Foxp3+ Tregs induce iNOS expression in myeloid suppressor cells (MSC). (B) CD4+/CD25-/Foxp3+ Tregs express iNOS and release large amounts of NO. NO from either MSC (A) or iNOS-expressing Tregs (B) is able to modulate the effector activity of CD4+/CD25-IFoxp3Th1 cells by reducing IL-2R signaling (STAT-5 activity) and IL-2 production. (C) Generation and activity of NO-Tregs. Large amounts of NO from inflammatory foci generate CD4+/CD25+/Foxp3-T cells, which activate p53 to increase IL2 production and proliferation. Similarly to CD4+/CD25+/Foxp3+ Tregs, NO-Tregs exert suppressive functions mainly through the production of IL-10.
During the development of T cells, auto-reactive CD4+CD8+ thymocytes are selected for deletion, and this negative selection occurs through activation-induced apoptosis of immature thymocytes(118). As an important pro-apoptotic agent, NO might play decisive roles in T cell selection and development in the thymus (Figure 4). This possibility is supported by the finding that IFN-β-deficient mice, which produce less iNOS-derived NO than their wild-type counterparts, show defective negative selection(119). In the thymus, iNOS is constitutively expressed by epithelial and dendritic cells of the corticomedullary junction and medulla, and is up-regulated after cell-cell interaction with thymocytes activated with auto- or allo-antigens(120,121). TCR-stimulated CD4+CD8+ thymocytes are highly sensitive to NO-mediated apoptosis, whereas CD4+CD8- and CD4-CD8+ thymocytes are not(120–122). Contrasting with this picture, a study in iNOS-deficient mice did not find significant differences from wild- type animals in the selection of CD8+ and CD4+ T lymphocytes, but the absence of iNOS rather reduced the development of thymic lymphomas in a tumorigenic p53 deficient background(123). Collectively, these studies suggest that NO from iNOS-positive thymic stromal cells might be one of the main inducers of TCR-mediated apoptosis, but that other factors must also be important. In this regard, it has been reported that mouse thymocytes can, in addition to iNOS, express nNOS and/or eNOS and produce low levels of Ca2+ induced NO even in the presence of lipopolysaccharide (LPS) and proinflammatory cytokines(84,89,124,125). Thus, NO generated by constitutive NOS in rat thymocytes might also play an important role in modulating thymocyte maturation(84,124). Regarding mechanism, formation of peroxynitrite and consequent protein nitration on tyrosine residues has emerged as an important inducer of apoptosis in immature thymocytes(125), similar to the situation described for activation-induced cell death in mature T lymphocytes(126).
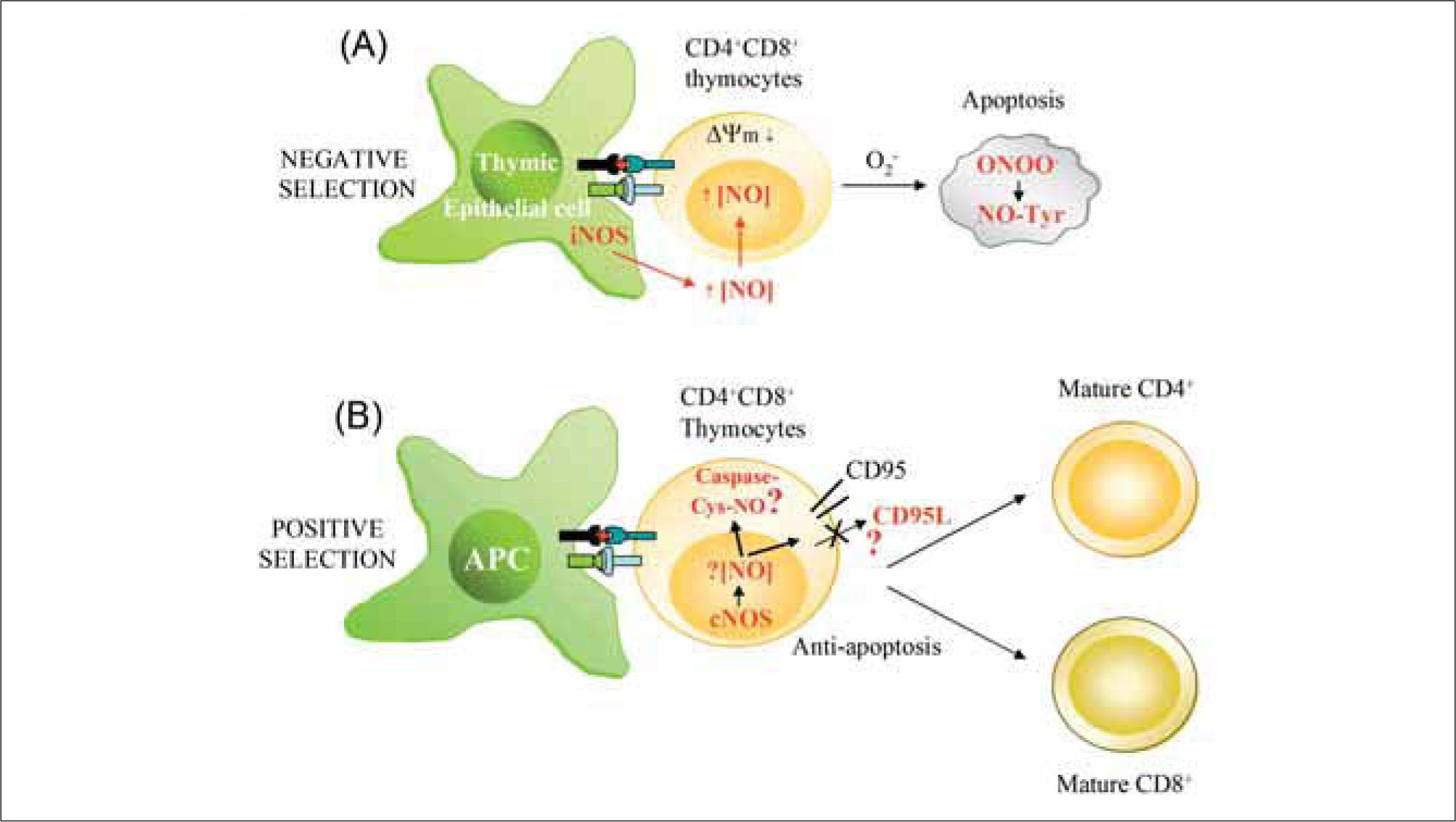
 Local high extracellular levels of NO produced by iNOS in thymic epithelial APCs promote negative selection of CD4+CD8+ thymocytes. iNOS-derived NO reduces mitochondrial membrane potential (¿ψm), generates ROS (O2-), and promotes the synthesis of peroxynitrite (ONOO-), which can induce apoptosis by nitration of proteins on Tyr (NO-Tyr). (B) Low levels of NO generated in T lymphocytes by constitutive NOS (nNOS and eNOS) may protect thymocytes from cell death during positive selection. nNOS/eNOS-derived NO may induce nitrosylation of caspases and/or reduce CD95L expression, thus favoring selection and proliferation of antigen-specific single positive thymocytes.")
Influence of NO on T cell development. (A) Local high extracellular levels of NO produced by iNOS in thymic epithelial APCs promote negative selection of CD4+CD8+ thymocytes. iNOS-derived NO reduces mitochondrial membrane potential (¿ψm), generates ROS (O2-), and promotes the synthesis of peroxynitrite (ONOO-), which can induce apoptosis by nitration of proteins on Tyr (NO-Tyr). (B) Low levels of NO generated in T lymphocytes by constitutive NOS (nNOS and eNOS) may protect thymocytes from cell death during positive selection. nNOS/eNOS-derived NO may induce nitrosylation of caspases and/or reduce CD95L expression, thus favoring selection and proliferation of antigen-specific single positive thymocytes.
Over the past two decades, there have been remarkable advances in the understanding of how NO can regulate immune responses. iNOS-expressing macrophages have historically been considered as the major source of NO and T lymphocytes the most important target cells. However, recent evidence indicates that constitutively expressed NOS in T lymphocytes also plays an important role in the development of adaptive immune responses. Overexpression of eNOS reduces the susceptibility of sensitized mice to allergic asthma upon challenge with ovalbumin(127), and the presence of eNOS, but not iNOS, is critical for anaphylactic shock(128,129). Moreover, antigen-dependent activation of eNOS regulates TCR signaling at the immune synapse by increasing the activation of CD3ζ, ZAP-70, and ERK1/2(85). The role of constitutive NOS expressed by T lymphocytes during adaptive immunity and the differentiation of Th17 polarized responses deserves further investigation.
ACKNOWLEDGMENTSWe thank Dr. P. Martin, K. McCreath, S. Cadenas, and S. Hortelano for critical readings and Dr. S. Bartlett for editorial assistance. This work was supported by grants PI070356 and Contrato-Investigador FIS to JMS (CP02/23024). S.I. holds a fellowship from CNIC-Bancaja.
ABBREVIATIONSNO (nitric oxide), APC (antigen-presenting cell), TCR (T cell receptor), NOS (nitric oxide synthase), MHP (mitochondrial hyperpolarization), MAPK (mitogen-activated protein kinases), ERK (extracellular signal-regulated kinase).
DISCLOSURESThe authors declare no competing financial interests.