




La presencia de lesión en el parénquima celular es común a un gran número de enfermedades crónicas del hígado, como por ejemplo las hepatitis virales, la hepatitis alcohólica, las colestasis crónicas o la esteatohepatitis. Aunque la patogenia puede variar según el agente etiológico, hay una serie de mecanismos comunes a todas ellas. Entre estos mecanismos destacan la activación de las células de Kupffer y el reclutamiento de células inflamatorias, la formación de radicales libres del oxígeno y la aparición de estrés oxidativo, la producción de citocinas, principalmente del factor de necrosis tumoral alfa y el factor de crecimiento transformante beta, y la liberación de mediadores de inflamación derivados de la oxidación del ácido araquidónico a través de la ciclooxigenasa 2 y la 5-lipooxigenasa.
The presence of a lesion in the cellular parenchyma is common to a large number of chronic liver diseases, such as viral hepatitides, alcoholic hepatitis, chronic cholestasis and steatohepatitis. Although the pathogenesis may vary according to the etiological agent, a series of mechanisms is common to all. Notable among these mechanisms are Kupffer cell activation and inflammatory cell recruitment, free oxygen radical formation and the development of oxidative stress, cytokine production, mainly TNFa and TGFb, and inflammatory mediator release due to arachidonic acid oxidation through the COX-2 and 5-LO pathways.
En todos los órganos y tejidos de nuestro organismo se producen episodios de lesión celular en respuesta a la acción de noxas físicas, químicas o biológicas (virus). En el caso concreto del hígado, la lesión celular se manifiesta principalmente con la muerte de las células parenquimatosas o hepatocitos. En el caso de que el daño hepático sea limitado, por ejemplo tras una hepatitis aguda, se produce una rápida respuesta regenerativa de los hepatocitos que reemplaza al tejido afectado y restablece la arquitectura hepática normal. Sin embargo, cuando el agente lesivo actúa de forma persistente y su acción sobrepasa la capacidad hepática de defensa y reparación, se produce una respuesta caracterizada por una regeneración celular desordenada y el desarrollo de inflamación y fibrosis1,2. Durante este proceso, la capacidad de regeneración hepática disminuye, mientras que la producción de componentes de la matriz extracelular (colágeno fibrilar, fibronectina, glucosaminoglicanos…) aumenta considerablemente. En fases avanzadas, la población normal de hepatocitos es parcialmente sustituida por la deposición desorganizada de estos componentes de la matriz extracelular, lo que causa una paulatina disminución de la masa hepatocelular y una progresiva distorsión anatómica y funcional del lobulillo hepático1,2. El resultado final es la cirrosis, desde el punto de vista anatómico, y la insuficiencia hepática crónica, desde el punto de vista funcional, una afección clínica que ocasiona la mayor parte de la morbimortalidad en las enfermedades hepáticas.
La aparición de daño hepatocelular es un hallazgo común a prácticamente todas las hepatopatías crónicas, como es el caso de la infección crónica por el virus de la hepatitis C, la hepatitis alcohólica, las colestasis crónicas o la esteatohepatitis no alcohólica1,2. De todas formas, aunque el desarrollo de lesión hepatocelular es común a la mayoría de enfermedades crónicas del hígado, su patogenia varía según el agente etiológico. Así, el daño hepático de origen alcohólico se caracteriza por la presencia de valores elevados de lipopolisacárido (LPS) de origen bacteriano con capacidad de promover la liberación de cantidades ingentes de agentes citotóxicos y de mediadores de inflamación (citocinas, eicosanoides…) por las células de Kupffer y de inducir la infiltración de leucocitos polimorfonucleares3. El daño hepático de origen alcohólico se produce también como consecuencia de la presencia de acetaldehído, que es el metabolito del alcohol con mayor capacidad para generar estrés oxidativo en los hepatocitos3. En el caso de las colestasis crónicas, el daño hepatocelular se debe a la acumulación de un exceso de ácidos biliares que estimulan la producción de radicales libres y desencadenan una intensa respuesta inflamatoria4. En el caso de las hepatopatías virales, la lesión hepatocelular está mediada por la respuesta inmune del huésped a los hepatocitos infectados que expresan en su membrana proteínas virales, lo que causa un alto grado de necrosis hepatocitaria y una reacción inflamatoria caracterizada por la infiltración de linfocitos5. Por último, en el caso de la esteatohepatitis no alcohólica, la manifestación hepática del síndrome metabólico, la acumulación de lípidos en el interior de los hepatocitos promueve el desarrollo de estrés oxidativo, inflamación, muerte celular y fibrosis6.
Hasta el momento se han descrito diversos mecanismos moleculares que son comunes a una gran parte de estas hepatopatías. Aunque la mayoría de estos mecanismos se han identificado en estudios realizados en modelos experimentales, los datos aportados se pueden extrapolar, con ciertas limitaciones, a la patología humana. Entre los mecanismos implicados en el desarrollo de lesión hepática destacan las siguientes: la activación de las células de Kupffer y el reclutamiento de células inflamatorias (neutrófilos y monocitos); la aparición de estrés oxidativo debido a la formación de radicales libres, principalmente derivados del oxígeno; la producción de citocinas, como el factor de necrosis tumoral alfa (TNFα) y el factor de crecimiento transformante beta (TGFβ) y, por último, la liberación de mediadores lipídicos de inflamación derivados del ácido araquidónico, como las prostaglandinas (PG), los leucotrienos (LT) y el factor de activación de las plaquetas (PAF). A continuación se describen con mayor detalle cada uno de estos mecanismos, haciendo especial hincapié en el papel en la necroinflamación y la fibrosis hepática de los mediadores lipídicos derivados del ácido araquidónico.
PAPEL DE LAS CÉLULAS DE KUPFFER Y DEL INFILTRADO INFLAMATORIOHay evidencias sólidas que demuestran la participación directa de las células de Kupffer en la patogenia de la lesión hepatocelular. Las células de Kupffer son los macrófagos residentes en el hígado y se encuentran formando parte de la unidad estructural funcional de este órgano, el sinusoide hepático7. El principal papel fisiológico de las células de Kupffer es eliminar mediante fagocitosis todo tipo de partículas extrañas, innecesarias o alteradas de la circulación sanguínea8. Por ejemplo, estas células desempeñan un papel clave en el proceso de captación y detoxificación de la endotoxina que llega del flujo venoso portal9,10. Las células de Kupffer actúan también, al igual que otros fagocitos mononucleares, como células presentadoras de antígeno que activan la respuesta inmune derivada de los linfocitos T11.
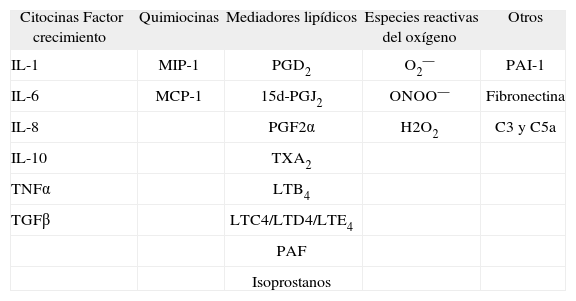
Se ha descrito que ciertos estímulos, como la obesidad, el consumo crónico de alcohol, la endotoxina o los compuestos procedentes de la degradación de fármacos o xenobióticos inducen la activación de las células de Kupffer12. Una vez activadas, estas células liberan cantidades ingentes de citocinas (interleucina [IL] 1, IL-6, IL-10 y TNFα), radicales libres del oxígeno (anión superóxido) y metabolitos del ácido araquidónico (PGD2, tromboxano [TX] A2, PGE2 y LT) (tabla I). En la mayoría de casos, la activación de las células de Kupffer se asocia al reclutamiento de neutrófilos y monocitos circulantes, los cuales amplifican de forma significativa la respuesta inflamatoria. En cualquier caso, la lesión tisular se produce cuando los mediadores de inflamación liberados por las células inflamatorias actúan de forma paracrina sobre las células adyacentes, ya sean hepatocitos o células hepáticas estrelladas12–15. La liberación desproporcionada de mediadores junto con la secreción de enzimas lisosomales por las células de Kupffer favorece la aparición de daño tisular, caracterizado principalmente por focos de necrosis y de inflamación. Las células de Kupffer activadas modifican también los procesos metabólicos clave en las células parenquimatosas, como la liberación de albúmina por los hepatocitos, lo que altera la función hepática y contribuye al desarrollo de la lesión en este órgano15. La prueba de concepto del papel que ejercen las células de Kupffer en la lesión hepática es que su inactivación y eliminación selectiva, mediante por ejemplo la administración de cloruro de gadolinio (GdCl3), normaliza los valores séricos de aspartato aminotransferasa (AST) y previene la lesión hepática tras la administración de tetracloruro de carbono (CCl4)16.
Principales mediadores de inflamación y lesión hepatocelular liberados por las células de Kupffer
| Citocinas Factor crecimiento | Quimiocinas | Mediadores lipídicos | Especies reactivas del oxígeno | Otros |
| IL-1 | MIP-1 | PGD2 | O2— | PAI-1 |
| IL-6 | MCP-1 | 15d-PGJ2 | ONOO— | Fibronectina |
| IL-8 | PGF2α | H2O2 | C3 y C5a | |
| IL-10 | TXA2 | |||
| TNFα | LTB4 | |||
| TGFβ | LTC4/LTD4/LTE4 | |||
| PAF | ||||
| Isoprostanos |
C3 y C5a: factores de complemento; H2O2: peróxido de hidrógeno; IL: interleucina; LT: leucotrieno; MIP-1: proteína inflamatoria de macrófagos-1; MCP-1: proteína quimioatrayente de monocitos-1; O2 : anión superóxido; ONOO : anión peroxinitrito; PAI-1: activador del plasminógeno; PAF: factor de activación plaquetaria; PG: prostaglandina; TGF: factor de crecimiento transformante; TNF: factor de necrosis tumoral; TX: tromboxano
Quizá el ejemplo paradigmático de la participación de las células de Kupffer en el daño hepático sea la hepatitis alcohólica. Es bien sabido que la ingesta abusiva de alcohol es capaz de alterar la permeabilidad intestinal y aumentar de esta forma la entrada al hígado de endotoxina a través de la vena porta. En condiciones normales, las células de Kupffer presentan un estado de tolerancia a la endotoxina, pero su exposición a cantidades elevadas de esta sustancia, o bien la presencia de células de Kupffer en estado parcialmente activado por otras causas, conduce a la liberación masiva de especies reactivas del oxígeno, citocinas (principalmente TNFα) y eicosanoides (principalmente LT) con capacidad de dañar el parénquima hepático17–19. También se ha apuntado que las células de Kupffer se activarían por factores liberados por los hepatocitos en respuesta al alcohol. Por ejemplo, se ha descrito que las células de Kupffer en presencia de un medio condicionado procedente de cultivos de hepatocitos tratados con alcohol, secretan IL-8, un potente agente quimiotáctico inductor de la infiltración de neutrófilos20. Otros autores, sin embargo, han demostrado que la liberación de IL-8 por los hepatocitos se produce en respuesta a factores derivados de las células de Kupffer activadas por el etanol, principalmente de IL-1 y TNFα21,22.
Otro ejemplo de la participación de las células de Kupffer en la lesión hepatocelular es el daño inducido por fármacos o xenobióticos. La vía hepatobiliar junto con el riñón constituyen la principal vía excretora de xenobióticos. Esto significa que las células hepáticas no sólo metabolizan los nutrientes procedentes del intestino y los productos metabólicos endógenos procedentes de otras partes del organismo, sino que también participan activamente en el metabolismo de los xenobióticos y fármacos. En condiciones normales, los fármacos son eliminados por la bilis primaria, hepatocelular o canalicular, elaborada por los hepatocitos mediante un proceso de filtración osmótica23. Sin embargo, en determinadas circunstancias, por ejemplo tras una sobredosis de fármacos, las células de Kupffer se activan y desencadenan un proceso de lesión hepática. Un ejemplo paradigmático de daño hepático inducido por fármacos lo constituye la intoxicación por acetaminofeno, que se caracteriza por la rápida aparición de insuficiencia hepática seguida por una intensa inflamación y, finalmente, regeneración del tejido lesionado24. La hepatotoxicidad al acetaminofeno se debe a la conversión por enzimas microsomales de este fármaco a un metabolito reactivo (la imina de N-acetil-p-benzoquinona), que se une a macromoléculas del tejido hepático e inicia una reacción necrótica y la activación de las células de Kupffer25.
La activación de las células de Kupffer se asocia habitualmente a la infiltración de células inflamatorias en el parénquima hepático. Esto es muy evidente en el caso del daño hepático por isquemia-reperfusión. Hay evidencias morfológicas de la activación de las células de Kupffer 1- 3 h después de la reperfusión del hígado26. Como consecuencia de su activación se produce el reclutamiento de neutrófilos y la liberación por ambos tipos celulares de anión superóxido, TNFα, factor activador de las plaquetas (PDGF), LTB4, PGD2, PGE2 y TXA2, factores que desencadenarían la lesión tisular27. La inactivación de las células de Kupffer mediante GdCl3 es capaz de reducir la lesión hepatocelular en modelos experimentales de reperfusión y trasplante27.
CITOCINAS Y FACTORES DE CRECIMIENTOLas citocinas y los factores de crecimiento son mediadores de comunicación celular de bajo peso molecular28. Aunque con notables diferencias en cuanto a la cantidad, casi todas las células de nuestro organismo, entre las que se incluye la mayoría de células hepáticas, poseen la capacidad de producir y secretar citocinas. Una vez liberadas, las citocinas interaccionan con receptores específicos en las células diana donde inducen múltiples respuestas, frecuentemente de una forma pleiotrópica28. El grupo de citocinas mas relevante son las IL (hay un total de 18 IL diferentes), el TNFα, el interferón gamma (IFNγ) y el TGFβ. En el contexto de la lesión hepática, el TNFβ y el TGFα desempeñan un destacado papel, ya que son importantes mediadores de inflamación y de remodelado de matriz extracelular y de fibrogénesis.
Hay amplias evidencias que indican la asociación entre la señalización del TNFα con la muerte hepatocelular29. Los efectos biológicos del TNFα están mediados por su unión a dos receptores de membrana: TNF-R1 y TNFR229. Los efectos biológicos del TNFα pueden estar también mediados por el factor de transcripción NF- κB29,30. En hepatocitos, la unión del ligando al receptor TNF-R1 se asocia a la inducción de apoptosis, aunque la participación del receptor TNF-R2 en la apoptosis mediada por células T no se puede descartar totalmente29,30. Además, en la mayoría de circunstancias, el TNFα induce la liberación de otras citocinas inflamatorias, como la IL-1 y la IL-6, por los macrófagos hepáticos, lo que amplifica y cronifica el grado de lesión hepatocelular29,30. En estudios realizados en humanos se ha descrito una estrecha asociación genética entre diversos polimorfismos del promotor del TNFα y el riesgo de desarrollar hepatopatía alcohólica31 y hepatitis fulminante32. En estudios experimentales se ha demostrado que el TNFα tiene un papel causal en la lesión hepática provocada por el alcohol y en la hepatotoxicidad inducida por Dgalactosamina33. En el modelo de D-galactosamina, por ejemplo, el TNFα induce la activación de la vía de las caspasas, lo que provoca la muerte de los hepatocitos por apoptosis, la infiltración de leucocitos y la muerte por fallo hepático33. Estos efectos hepatotóxicos estarían mediados por el receptor TNF-R1, puesto que los ratones deficientes para este receptor están protegidos frente al daño hepático inducido por la D-galactosamina34. Además, el TNFα ejerce un papel importante en el modelo de CCl4, ya que los ratones deficientes para sus 2 receptores son resistentes al desarrollo de necroinflamación y fibrosis inducido por este hepatotóxico35.
Evitar los efectos dañinos del TNFα en los hepatocitos proporcionaría una vía terapéutica importante para el tratamiento de la hepatopatía alcohólica o tóxica. Una de las alternativas terapéuticas disponibles actualmente es el fármaco infliximab (Remicade®), un anticuerpo monoclonal quimérico dirigido contra TNFα. Este anticuerpo se utiliza actualmente en el tratamiento de la enfermedad de Crohn y la artritis reumatoide, y está en fase de ensayo clínico para el tratamiento de la sarcoi dosis y la vasculitis36–38. En el caso del hígado, dos estudios clínicos preliminares apuntan hacia un efecto beneficioso del infliximab en pacientes con hepatitis alcohólica grave39,40. Sin embargo, se han documentado la aparición de efectos adversos (p. ej., mayor incidencia de infecciones) y los ensayos con infliximab han sido suspendidos41. Además, la seguridad de este fármaco se halla en entredicho, sobre todo después de haberse detectado episodios de reactivación del virus de la hepatitis B o de la hepatitis autoinmune en pacientes con enfermedad de Crohn o con artritis bajo tratamiento con este fármaco42,43.
El TGFβ es una citocina/factor de crecimiento involucrada en la progresión de la lesión hepática. El TGFβ es esencialmente un citocina profibrótica que desempeña un papel fundamental en la activación de las células hepáticas estrelladas (HSC) y en el desarrollo de fibrogénesis44. En concreto, en respuesta al TGFβ liberado por las células de Kupffer, las HSC sufren un proceso de transdiferenciación desde un fenotipo quiescente almacenador de lípidos, a un fenotipo activado que se asemeja a un miofibroblasto. Una vez activadas, las HSC producen los principales componentes de la matriz extracelular, y secretan mediadores proinflamatorios (citocinas, quimiocinas…), los cuales activan las HSC circundantes y, por tanto, autoperpetúan el daño hepático. De hecho, algunos estudios experimentales demuestran que los ratones transgénicos que sobreexpresan TGFβ desarrollan fibrosis hepática de forma espontánea45, mientras que el bloqueo de su síntesis o de su mecanismo de señalización da lugar a un efecto antifibrogénico neto46.
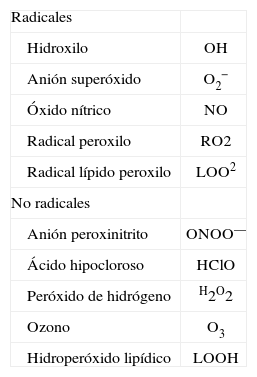
ESTRÉS OXIDATIVOLa mayoría de procesos biológicos relacionados con la generación de energía por la mitocondria y las reacciones de detoxificación celular generan de forma inevitable radicales libres, moléculas altamente inestables que contienen un electrón desapareado y, por tanto, altamente reactivo47. Hay factores externos que aumentan la producción de radicales libres en nuestro organismo, como el consumo de tabaco y alcohol o las radiaciones ionizantes47. Los radicales libres más comunes son los productos derivados del metabolismo del oxígeno (espe- cies reactivas del oxígeno [ROS]) (tabla II) y del nitrógeno (especies reactivas del nitrógeno [RNS]). ROS y RNS pueden combinarse entre ellos dando lugar a especies más tóxicas o dañinas, como el anión peroxinitrito (ONOO—), un producto resultante de la reacción del anión superóxido (O2 —) y el óxido nítrico (NO). En el organismo, las ROS reaccionan ávidamente con sustratos orgánicos, como los lípidos, las proteínas y el ADN, dañando las células y promoviendo el desarrollo de diversas enfermedades47. Afortunadamente, nuestro organismo posee sistemas antioxidantes, como la superóxido dismutasa (SOD) o la glutatión peroxidasa, con capacidad de amortiguar los efectos de las ROS. En condiciones normales, el balance entre las ROS y los sistemas antioxidantes está equilibrado, pero en ciertas circunstancias puede haber una sobreproducción de ROS o un descenso de los sistemas antioxidantes, dando lugar a la aparición de estrés oxidativo y lesión celular47.
Lista de las especies reactivas del oxígeno más relevantes en la lesión hepática
| Radicales | |
| Hidroxilo | OH |
| Anión superóxido | O2− |
| Óxido nítrico | NO |
| Radical peroxilo | RO2 |
| Radical lípido peroxilo | LOO2 |
| No radicales | |
| Anión peroxinitrito | ONOO— |
| Ácido hipocloroso | HClO |
| Peróxido de hidrógeno | H2O2 |
| Ozono | O3 |
| Hidroperóxido lipídico | LOOH |
El estrés oxidativo ejerce un papel preponderante en la patogenia de la lesión hepatocelular. De hecho, se ha descrito una sobreproducción de ROS y un aumento de la peroxidación lipídica en pacientes con hepatitis alcohólica, esteatohepatitis y cirrosis48. De forma experimental, hay estudios que apoyan un papel central para el estrés oxidativo en la patogenia de la lesión hepática. En concreto, en el modelo de CCl4, en el cual la generación de radicales libres produce una lipoperoxidación hepática masiva, la administración de agentes antioxidantes, como el cinc, la vitamina E o el S-adenosil-metionina (SAME), es capaz de contrarrestar y atenuar el daño hepatocelular12.
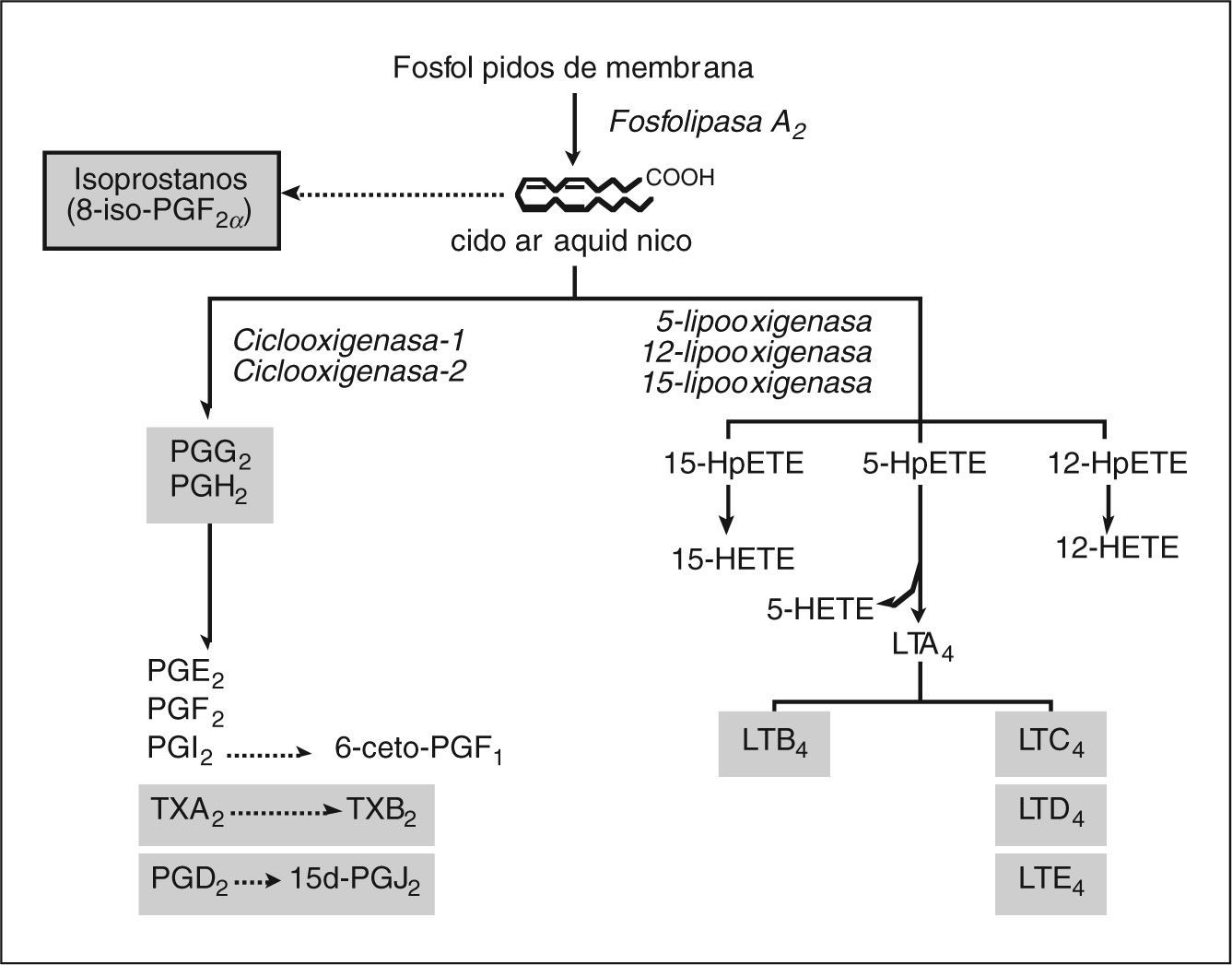
MEDIADORES LIPÍDICOS DE INFLAMACIÓNLos mediadores lipídicos de inflamación son compuestos de naturaleza lipídica con bajo peso molecular y una gran actividad biológica. Representan una de las redes de comunicación celular más complejas del organismo. La biosíntesis de mediadores lipídicos se inicia generalmente en respuesta a un estímulo que induce a las células a utilizar los lípidos de su membrana celular para generar mediadores lipídicos que actúan como señales intracelulares o extracelulares. Un ejemplo paradigmático de esta clase de mediadores son los metabolitos del ácido araquidónico, un ácido graso poliinsaturado que se libera de los fosfolípidos de la membrana celular plasmática mediante la acción de la enzima fosfolipasa A249. Alternativamente, el pool de fosfolípidos de membrana puede ser metabolizado a 1-alquil-2-acetil-lisofosfatidilcolina o PAF, otro potente mediador lipídico50. Las concentraciones intracelulares de ácido araquidónico libre en células de mamíferos son normalmente muy bajas, ya que este ácido graso es rápidamente metabolizado a un grupo de compuestos que se denominan de forma genérica eicosanoides. En los mamíferos hay dos principales vías de metabolismo del ácido araquidónico: la vía de la ciclooxigenasa (COX) y la vía de las lipooxigenasas (LO), mediante las cuales se sintetizan la mayor parte de los eicosanoides biológicamente activos, como los prostanoides (PG y TX), los LT y los ácidos hidroxieicosatetraenoicos (HETE) (fig. 1)51,52.
 y las lipooxigenasas (LO), o bien de forma no enzimática mediante la acción de los radicales libres a isoprostanos. Mediante la vía de la COX, de la cual hay dos isoformas (COX-1 y COX-2), el ácido araquidónico es transformado a las distintas prostaglandinas (PG) y tromboxano (TX) A2. La PGD2 es convertida de forma no enzimática a la 15- deoxi-PGJ2 (15d-PGJ2). Mediante la vía de las LO, la cual incluye la 5-, la 12- y la 15- LO, el ácido araquidónico da lugar a los ácidos hidroperoxieicosatetraenóicos 5-, 12-y 15-HpETE, que son posteriormente reducidos a 5-, 12- y 15-HETE, respectivamente. El 5-HpHETE también puede ser hidrolizado a LTB4 o bien convertido en LTC4/LTD4/ LTE4, los cisteinil-LT. En este esquema se resaltan en un cuadro sombreado los mediadores derivados del ácido araquidónico con implicación directa en el daño hepatocelular.")
Mediadores lipídicos de inflamación derivados del ácido araquidónico. Los fosfolípidos presentes en la membrana celular son transformados a un ácido graso de 20 átomos de carbono, el ácido araquidónico, el cual es oxidado de forma enzimática por las vías de la ciclooxigenasa (COX) y las lipooxigenasas (LO), o bien de forma no enzimática mediante la acción de los radicales libres a isoprostanos. Mediante la vía de la COX, de la cual hay dos isoformas (COX-1 y COX-2), el ácido araquidónico es transformado a las distintas prostaglandinas (PG) y tromboxano (TX) A2. La PGD2 es convertida de forma no enzimática a la 15- deoxi-PGJ2 (15d-PGJ2). Mediante la vía de las LO, la cual incluye la 5-, la 12- y la 15- LO, el ácido araquidónico da lugar a los ácidos hidroperoxieicosatetraenóicos 5-, 12-y 15-HpETE, que son posteriormente reducidos a 5-, 12- y 15-HETE, respectivamente. El 5-HpHETE también puede ser hidrolizado a LTB4 o bien convertido en LTC4/LTD4/ LTE4, los cisteinil-LT. En este esquema se resaltan en un cuadro sombreado los mediadores derivados del ácido araquidónico con implicación directa en el daño hepatocelular.
De forma clásica, se conoce la existencia de la vía de la COX, mediante la cual el ácido araquidónico es convertido a las distintas PG y TX50,51. La COX es una hemoproteína de membrana formada por dos polipéptidos idénticos de 70 kD, que funciona como una enzima bifuncional llevando a cabo las dos primeras etapas de la biosíntesis de prostanoides: a) la oxidación del ácido araquidónico para formar la estructura cíclica PGG2 (actividad ciclooxigenasa propiamente dicha), y b) la peroxidación de la PGG2 para originar el PGH2 (actividad peroxidasa)50,51. Hasta el momento, se han clonado y caracterizado dos isoformas de la COX: COX-1 y COX-2. Aunque ambas isoformas son sensibles en mayor o menor grado a la inhibición por parte de los antiinflamatorios no esteroideos, estas isoformas se distinguen esencialmente por su función fisiológica. En este sentido, se considera que la COX-1 es una isoforma constitutiva que tendría un papel clave en el mantenimiento de las funciones homeostáticas básicas, promovería la producción de PG básicamente protectoras y sería la forma predominante en la mucosa gástrica y las plaquetas53. Por el contrario, y con notables excepciones, como el riñón y el sistema nervioso, la COX-2 es una isoforma inducible que no se expresa en condiciones normales; su expresión se induce en procesos inflamatorios y de proliferación, y diferenciación celular53. En cualquier caso, independientemente de su origen, el endoperóxido PGH2 constituye el metabolito intermediario de la biosíntesis de las distintas PG: PGE2, PGI2 o prostaciclina, PGF2α y PGD2 por sintasas terminales específicas. Así, por ejemplo, la PGH2 es transformada por la PGD sintasa a PGD2, por la PGE sintasa a PGE2, por la PGF sintasa a PGF2α y por la PGI sintasa a PGI2 o prostaciclina. Se han descrito tres PGE sintasas distintas (cPGE, mPGE-1 y mPGE-2) y dos isoformas de la PGD sintasa (H-PGD y L-PGD). Sobre la PGH2 actúa también la TX sintasa que origina el TXA2. Tanto PGI2 como TXA2 son compuestos muy inestables (con una vida media de 3 min para la PGI2 y de 30 s para el TXA2) y se hidrolizan espontáneamente a 6-ceto-PGF1α y TXB2, respectivamente, los cuales no poseen actividad biológica. Asimismo, la PGD2 es convertida de forma no enzimática a la 15-deoxi-PGJ2 (15d-PGJ2), que sí posee actividad biológica (ver más adelante). Hasta el momento se han descrito 10 diferentes tipos y subtipos de receptores de prostanoides, los cuales al igual que las sintasas responsables de su síntesis, se expresan de forma específica en cada uno de los tejidos y tipos celulares54,55. Cuatro de estos receptores reconocen a la PGE2 (EP1, EP2, EP3 y EP4), 2 unen PGD2 (DP1 y DP2), 2 unen TXA2 (TPα y TPβ) y los 2 restantes son receptores únicos para PGF2α y PGI2 (FP y IP, respectivamente)54,55. Los prostanoides, especialmente las PG y, entre ellas, la PGE2, desempeñan un papel fundamental en la inflamación, de forma que la inhibición de su síntesis representa el mecanismo de acción establecido de los fármacos antiinflamatorios actualmente disponibles56.
La segunda vía de metabolismo del ácido araquidónico es la vía de las LO, la cual agrupa a una familia de dioxigenasas que transforman el ácido araquidónico en LT y HETE51,52. En humanos hay tres distintas LO, que se denominan 5-LO, 12-LO y 15-LO, las cuales incorporan una molécula de oxígeno en los carbonos 5, 12 o 15, res- activapectivamente, del ácido araquidónico51,52. La 5-LO convierte el ácido araquidónico en 5-HETE y LT, mientras que la 12-LO y la 15-LO generan los correspondientes 12- y 15-HETE, respectivamente51,52. La vía de la 5-LO es la más relevante desde el punto de vista fisiológico y farmacológico, ya que es la responsable de la síntesis de LT. Para sintetizar LT es necesaria la translocación de la 5-LO desde el citosol a la membrana nuclear, donde interacciona con la proteína activadora de la 5-LO (FLAP), la cual facilita la presentación del ácido araquidónico al centro activo de la 5-LO y la transformación de este en 5-HPETE51,52. El 5-HPETE es posteriormente convertido por la misma 5-LO a 5-HETE y a LTA451,52. El LTA4 es un compuesto inestable y altamente reactivo, que es rápidamente transformado por la LTA4 hidrolasa a LTB4, o bien conjugado con el glutatión mediante la actividad de la enzima LTC4 sintasa para dar lugar a LTC4. El LTC4 es transformado mediante pérdida sucesiva de residuos aminoacídicos en LTD4 y LTE4, los cuales reciben el nombre genérico de cisteinil-LT. Una vez sintetizados y liberados, los productos derivados de la 5-LO ejercen sus efectos biológicos mediante la activación de receptores acoplados a proteína G. Hasta el momento, se han clonado 2 receptores distintos para el LTB4 y 2 para los cisteinil- LT. Los receptores B-LT1 y B-LT2 reconocen al LTB4 con alta y baja afinidad, respectivamente. El receptor B-LT1 se localiza principalmente en los leucocitos y su activación induce una notoble respuesta quimiotáctica, mientras que el receptor B-LT2 se expresa en la mayoría de tejidos, aunque su función es actualmente desconocida55. Los dos receptores para los cisteinil-LT, Cys-LT1 y Cys-LT2, reconocen LTC4 y LTD4. El receptor Cys-LT1 se localiza abundantemente en el músculo liso pulmonar y su activación se asocia a vasoconstricción y adhesión celular55. El receptor Cys-LT2 se distribuye de forma uniforme entre las venas pulmonares, el bazo, las fibras de Purkinje, el corazón y la glándula adrenal, aunque su función es todavía desconocida55. Los LT son potentes mediadores de inflamación y daño celular57. El LTB4, en concreto, es un potente agente quimioatrayente, un potente inductor de adhesión celular y el principal estímulo para la producción de ión superóxido y la liberación de enzimas hidrolíticos por el neutrófilo57. Asimismo, los cisteinil-LT, que originalmente se identificaron como las sustancias de reacción lenta liberadas en el curso de las reacciones anafilácticas (slow reacting substance of anaphylaxis), además de su actividad vasoconstrictora, son potentes agentes quimioatrayentes para los eosinófilos, incrementan la permeabilidad vascular en las vénulas postcapilares e inducen la síntesis y la liberación de otros mediadores de inflamación, como la IL-8 y el PAF57.
Dado que la síntesis de eicosanoides se produce normalmente en células inflamatorias, como neutrófilos, eosinófilos, macrófagos y mastocitos, la célula de interés en el contexto del hígado sería la célula de Kupffer. De hecho, una vez activadas, las células de Kupffer, expresan COX- 1 y COX-2 y sintetizan la mayoría de productos de esta vía, entre las que se incluyen PGE2, PGD2, PGF2α, PGI2 y TXA215,58. El principal prostanoide liberado por las células de Kupffer activadas es la PGD258, el precursor de la 15d-PGJ2 (ver más adelante). Además, se ha descrito que la liberación de PGE2 por la célula de Kupffer en respuesta al alcohol induce la síntesis de triglicéridos en hepatocitos y contribuye a la aparición de esteatosis19. Respecto a la vía de la 5-LO, clásicamente se ha considerado a la célula de Kupffer como la responsable de la síntesis de LT en el hígado59. De hecho, en el tejido hepático, las células de Kupffer son el único tipo celular que posee la maquinaria enzimática necesaria (5-LO, FLAP, LTA4 hidrolasa y LTC4 sintasa) para la síntesis de estos eicosanoides60,61. La producción de LTB4 en el hígado parece depender casi exclusivamente de las células de Kupffer, aunque hay estudios realizados en hepatocitos en cultivo que indican que el alcohol puede estimular la liberación al medio de una sustancia con propiedades quimioatrayentes de estructura semejante al LTB462. Sin embargo, algunos estudios recientes demuestran que la producción de LTB4 por los hepatocitos es irrelevante, ya que no hay ninguna evidencia directa de la presencia de LTA4 hidrolasa en estas células63. Lo que no se puede descartar es que parte de los cisteinil-LT producidos en el hígado puedan originarse por un metabolismo transcelular, puesto que la LTC4 sintasa se expresa también en los hepatocitos y en las células endoteliales61. De hecho, en nuestro laboratorio hemos demostrado que el LTA4 producido por las células de Kupffer es transformado por los hepatocitos a cisteinil-LT61.
La participación de los mediadores lipídicos derivados del ácido araquidónico en la lesión hepática es un tema de interés creciente. En primer lugar, se ha demostrado que la expresión de la COX-2 se halla aumentada en pacientes infectados crónicamente con el virus de la hepatitis B o C y en los pacientes con cirrosis64–67. Además, en los pacientes con hepatitis C crónica hay una estrecha correlación entre el grado de progresión a fibrosis hepática y la expresión de la COX-264. La inducción de la COX-2 y el aumento de los valores hepáticos de PG se han confirmado experimentalmente en ratas en que se ha inducido una lesión hepática con CCl468, en modelos de hepatopatía alcohólica69,70 y en ratones con esteatohepatitis no alcohólica inducida por dietas lipogénicas deficientes en colina y metionina71–73. Un brillante estudio publicado recientemente demuestra que los ratones transgénicos que sobreexpresan el gen de la COX-2 humana en el hígado presentan valores elevados de transaminasas y signos histológicos de hepatitis74. En otro estudio reciente, se ha demostrado que el replicón del virus de la hepatitis C es capaz de estimular la expresión de la COX-2 y aumentar los valores de PGE2 en cultivos de líneas celulares de hepatocitos75. En células hepáticas estrelladas en cultivo, la expresión de la COX-2 se induce en respuesta a sales biliares y mitógenos, como el PDGF, los cuales aumentan la proliferación y la resistencia a la apoptosis en estas células76,77. Actualmente hay una cierta controversia respecto a si el efecto fibrogénico de la COX-2 está mediado o no por PG, ya que se han observado efectos reguladores tanto positivos como negativos de la PGE2 sobre la activa ción de las células hepáticas estrelladas en cultivo78–81. Resultados más concluyentes se han obtenido mediante el uso de inhibidores selectivos de la COX-2. Estos compuestos reducen la proliferación de las células hepáticas estrelladas en cultivo y ejercen efectos antifibrogénicos en modelos animales de esteatohepatitis o con fibrosis hepática inducida por CCl468,72,82–84. Resta por esclarecer si los efectos antifibrogénicos de los inhibidores de la COX- 2 están más bien mediados por la activación de PPARγ, que por la propia inhibición de la síntesis de PG85.
Efectos similares a la vía de la COX-2 se han descrito para los metabolitos de la vía de la 5-LO. En este sentido, se ha demostrado un incremento de la expresión de la 5-LO y un aumento de los valores hepáticos de metabolitos de la 5-LO en ratas con fibrosis hepática inducida por CCl4 o tioacetamida61,86,87. En línea con estos resultados, se ha demostrado un aumento de la excreción urinaria de productos derivados de la 5-LO en pacientes con enfermedad hepática crónica88,89. En estudios in vitro se ha demostrado que los productos de la 5-LO, principalmente el LTD4, son capaces de estimular la proliferación y la síntesis de colágeno en células hepáticas estrelladas y fibroblastos90–92. En modelos experimentales, el bloqueo de la vía de la 5-LO mediante el compuesto Bay-X-1005, un inhibidor de la FLAP, reduce la necroinflamación y la fibrogénesis hepática93,94. Efectos similares se han obtenido mediante un inhibidor de la 5-LO, el compuesto CJ- 13,61084. Además, se ha demostrado que los ratones deficientes (knockout) para la 5-LO están protegidos ante el daño necroinflamatorio inducido por el CCl484. Se ha sugerido que el mecanismo por el cual la inhibición de la 5- LO se asocia a un efecto antifibrogénico estaría relacionado no sólo con la inhibición de la síntesis de LT sino también a la inducción de apoptosis en las células inflamatorias del hígado93. De hecho, se ha demostrado que los inhibidores de la 5-LO inducen apoptosis en células de Kupffer en cultivo93 y que los efectos antifibrogénicos de estos compuestos se producen de forma paralela a un descenso significativo del número de macrófagos93,94. Puesto que los inhibidores de la 5-LO inducen apoptosis de forma exclusiva en células que contienen una vía de la 5-LO metabólicamente activa, y dado que en el hígado esto sólo ocurre en las células de Kupffer y en las células inflamatorias infiltradas61, la inhibición de la 5-LO representa una estrategia válida para la prevención de la inflamación y la fibrosis hepática. De hecho, la inhibición de la vía de la 5-LO y la modulación de la función de las células de Kupffer constituye el mecanismo de acción de flavonoides naturales, como la silimarina, cuya eficacia ha sido evaluada en pacientes con hepatitis alcohólica y esteatohepatitis no alcohólica95,96.
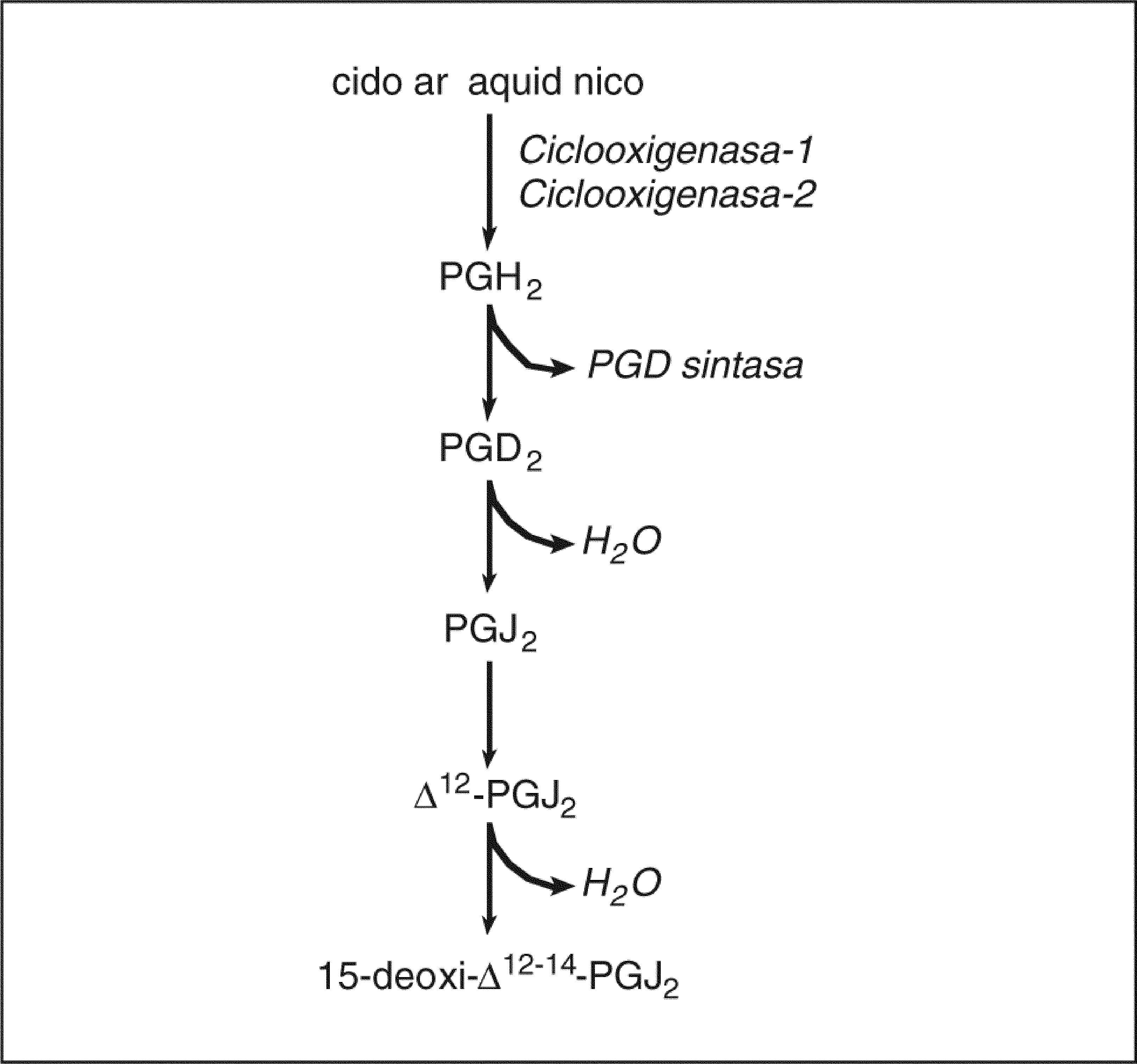
Prostaglandinas ciclopentenonasUn capítulo aparte lo constituyen las PG ciclopentenonas (cyPG), que son los productos no enzimáticos de la deshidratación de las PG (fig. 2). Las cyPG se caracterizan estructuralmente por la presencia de un grupo carbonilo no saturado altamente reactivo en el anillo ciclopentenona97. Las cyPG más relevantes desde el punto de vista biológico son las PG de la serie J2 (PGJ2, ¿12-PGJ2 y 15d-PGJ2), todas ellas derivadas de la PGD2. No se ha descrito hasta el momento ningún receptor que reconozca las cyPG, pero sí se ha demostrado que la 15d-PGJ2 es un ligando natural del PPARγ, cuya activación posiblemente medie las propiedades antiinflamatorias de esta CyPG98. De los efectos biológicos de las CyPG destacan principalmente las propiedades antiproliferativas y proapoptóticas mediadas por la modulación de H-Ras, MAPK y Akt99,100. Aparentemente, la fuente principal de 15d-PGJ2 es la isoforma COX-2. Nuestro laboratorio ha demostrado un aumento de los valores hepáticos de 15d-PGJ2 de forma paralela a la sobreexpresión de COX-2 durante la lesión hepática inducida por el CCl468. En este estudio, la inhibición de la COX-2 disminuyó significativamente la formación hepática de 15d-PGJ2 y confirió un estado de hepatoprotección. Estos resultados son consistentes con el hecho de que la 15d-PGJ2 aumenta el daño hepatocelular inducido por el alcohol alílico101.
 ciclopentenonas. La PGD2, que se genera mediante la acción secuencial de la ciclooxigenasa y la PGD sintasa, da lugar mediante mecanismos no enzimáticos de deshidratación a la PGJ2 ciclopentenona y sus derivados △12-PGJ2 y 15-deoxi-△12-14-PGJ2 (15d-PGJ2).")
Biosíntesis de prostaglandinas (PG) ciclopentenonas. La PGD2, que se genera mediante la acción secuencial de la ciclooxigenasa y la PGD sintasa, da lugar mediante mecanismos no enzimáticos de deshidratación a la PGJ2 ciclopentenona y sus derivados △12-PGJ2 y 15-deoxi-△12-14-PGJ2 (15d-PGJ2).
Se ha descrito la producción de ROS durante la oxidación del ácido araquidónico a través de la COX y las LO102. De hecho, los productos iniciales de la síntesis de eicosanoides generan metabolitos intermediarios que son también altamente peroxidativos. Además, algunos de estos intermediarios, como el LTA4, es capaz de formar aductos en el tejido. Otras reacciones mediadas por las LO generan peróxidos reactivos, como el ácido 13-hidroxioctadecadienóico (13-HODE) a partir de la 15-LO103. Por último, apuntar que algunas de las enzimas de la cascada del ácido araquidónico, como el glutatión S-transferasa o el epóxido hidrolasa, pertenecen a familias cuyas funciones están involucradas en la detoxificación celular. Esto sugiere que las vías de biosíntesis de eicosanoides podrían haber evolucionado originalmente a partir de los sistemas primitivos de detoxificación.
IsoprostanosLas membranas lipídicas son altamente susceptibles al ataque de los radicales libres del oxígeno y a la peroxidación. Así, el ácido araquidónico presente en los fosfolípidos de membrana sufre un proceso de peroxidación in situ que da lugar a la síntesis de isoprostanos. A diferencia de la producción de PG catalizada por la COX, los isoprostanos formados in situ se esterificarían en los lípidos tisulares y se liberarían subsecuentemente preformados104,105. Su biosíntesis incluye la formación de bicicloendoperóxidos intermediarios del tipo PGH2, que son reducidos a 4 regioisómeros del tipo PGF2α y que se les conoce colectivamente como F2 isoprostanos. El 8-iso- PGF2α es el isoprostano más abundante y constituye el estándar de referencia de la determinación in vivo del grado de estrés oxidativo106.
Dado que los isoprostanos son isómeros de las PG, se ha postulado que estos compuestos no son simples marcadores de peroxidación lipídica, sino que también poseen actividad biológica específica. En este sentido, se ha demostrado que el 8-iso-PGF2α es un potente vasoconstrictor, un potente activador de la proliferación de células musculares lisas y endoteliales, así como un potencial mediador de daño oxidativo107. Respecto a este último punto, se ha descrito que la formación de isoprostanos per se causa una clara distorsión de la membrana plasmática, dando lugar a cambios en su fluidez e integridad108. Además, se ha demostrado que la presencia de valores elevados de isoprostanos induce citotoxicidad en células en cultivo109. Respecto al hígado, la primera evidencia de la formación in vivo de F2-isoprostanos se obtuvo en el modelo de lesión hepática inducido por CCl4110. Posteriormente, se demostró la presencia de valores incrementados de F2-isoprostanos en la orina de pacientes con hepatitis alcohólica111 y en modelos experimentales de esta enfermedad112. Por último, se ha constatado que los pacientes con hepatitis C o cirrosis biliar primaria también presentan una excreción urinaria de F2-isoprostanos elevada113,114.
Efectos protectores de los ácidos grasos omega-3Al contrario de los ácidos grasos omega-6 (el ácido araquidónico, el precursor común de PG y LT, es uno de ellos), los ácidos grasos omega-3 poseen propiedades antiinflamatorias y hepatoprotectoras. Los efectos beneficiosos de los omega-3 estarían mediados por al menos cuatro mecanismos distintos: a) la inhibición de la disponibilidad de ácido araquidónico en la membrana celular plasmática para la síntesis de PG y LT; b) la inhibición directa de las vías de la COX-2 y la 5-LO; c) la síntesis a partir de los propios ácidos grasos omega-3 de PG de la serie 3 y LT de la serie 5 con menor capacidad inflamatoria que las PG de la serie 2 y LT de la serie 4, que son las que se producen a partir del ácido araquidónico, y d) la síntesis a partir de los propios ácidos grasos omega-3 de nuevos mediadores lipídicos con claras propiedades antiinflamatorias (p. ej., resolvinas y protectinas)115,116. En función de estos resultados, en nuestro laboratorio hemos demostrado que las dietas ricas en los ácidos grasos omega- 3 ácido docosahexaenóico y ácido eicosapentaenóico reducen de forma significativa la necroinflamación hepática en ratones con daño hepático inducido por CCl4117. Los efectos hepatoprotectores de los omega-3 serían en este caso secundarios a la inhibición de la síntesis de mediadores lipídicos derivados de las vías de la 5-LO y la COX-2, al descenso del daño genotóxico y de los valores de estrés oxidativo en los hepatocitos y a la disminución de la liberación de TNFα por los macrófagos117.
Nuestro laboratorio está financiado por el Ministerio de Educación y Ciencia (SAF 06/03191). CIBEREHD está financiado por el Instituto de Salud Carlos III.

